
奥美拉唑肠溶胶囊人体生物等效性临床研究
2024-10-10 00:00:00茅关兴赵丽敏李琦君徐诗强
上海医药 2024年15期
摘 要 评估自制奥美拉唑肠溶胶囊(20 mg)与参比制剂(LOSEC,20 mg)在体内的生物等效性。受试者均为健康成人,盲法随机服用自制制剂或参比制剂的奥美拉唑肠溶胶囊。通过测定血浆中奥美拉唑的浓度,以药代动力学参数为指标比较两种制剂的生物等效性,并评价两制剂体内安全性。结果显示,在空腹及餐后条件下,自制制剂与参比制剂的奥美拉唑药动学参数几何均数比值均落在生物等效性接受范围内,具有生物等效性,不良事件发生率相近,所有不良反应均为轻度,安全性无显著差异。
关键词 奥美拉唑 肠溶胶囊 一致性评价 生物等效性
中图分类号:R969.4; R975.2 文献标志码:A 文章编号:1006-1533(2024)15-0078-05
引用本文 茅关兴, 赵丽敏, 李琦君, 等. 奥美拉唑肠溶胶囊人体生物等效性临床研究[J]. 上海医药, 2024, 45(15): 78-82.
基金项目:上海市战略性新兴产业发展专项资金资助项目
Clinical studies on human bioequivalence of omeprazole enteric-coated capsules
MAO Guanxing1, ZHAO Limin2, LI Qijun3, XU Shiqiang4
(1. Shanghai Pharmaceutical Industry Co., Ltd., Shanghai 200002, China; 2. Shanghai Meiyou Pharmaceutical Co., Ltd., Shanghai 201422, China; 3. Shenyang Dasan Pharmaceutical Technology Co., Ltd., Shenyang 110179, China; 4. School of Medicine, Wuhan University of Science and Technology, Wuhan 430065, China)
ABSTRACT To evaluate the bioequivalence of generic omeprazole enteric-coated capsules (20 mg) versus a reference formulation (LOSEC, 20 mg) in vivo. Healthy adult subjects were enrolled and randomly and blindly administered with either the generic formulation or the reference formulation of omeprazole enteric-coated capsules. By measuring the concentration of omeprazole in plasma, the bioequivalence of the two formulations was compared using pharmacokinetic parameters and their in vivo safety was evaluated. The results showed that under fasting and postprandial conditions, the geometric mean ratio of pharmacokinetic parameters of the two preparations fell within the acceptance range of bioequivalence, the adverse reactions associated with generic preparation were mild and similar to those of reference formulation, and there were no distinctive differences in safety between the two preparations.
KEY WORDS omeprazole; enteric-coated capsules; consistency evaluation; bioequivalence
奥美拉唑(omeprazole)是瑞典Astra公司研发的质子泵抑制剂(PPI),主要用于治疗消化性溃疡及与胃酸有关的消化系统功能紊乱性疾病[1-2]。由于奥美拉唑在酸性溶液中易分解,为增加药物的稳定性,防止其被胃酸破坏,参比制剂采用肠溶包衣保证奥美拉唑的稳定性和生物利用度。肠溶制剂是一种在体内释放行为较为复杂的药物剂型,其研发过程在仿制药领域中具有较高的难度。肠溶制剂的释放行为受到多种因素的影响,如制剂的组成、结构、制备工艺以及体内环境等,尤其在餐后生物等效性试验中,由于进食后生理条件发生显著变化,肠溶制剂的释药行为及药代动力学特征会出现较大的变异,自制制剂存在生物不等效的风险[3]。本研究遵循国家一致性评价的标准与要求,开发和生产了自制奥美拉唑肠溶胶囊。为确保制剂的质量与疗效,我们对工艺验证批次的自制产品与参比制剂进行了人体生物等效性试验,以便全面验证自制制剂在临床应用中的疗效,确保其与参比制剂达到生物等效。
1 材料和方法
1.1 药品
奥美拉唑肠溶胶囊受试制剂(T)(规格20 mg/粒,上海美优制药有限公司);参比制剂(R)LOSEC(规格20 mg/粒,Astra Zeneca UK Limited)。
1.2 仪器
UPLC I-class 液相色谱仪、Xevo TQ-S 质谱仪(美国Waters公司);Hypersil GOLD C18 选择性 HPLC 色谱柱和预分析柱(2.1 mm×50 mm,1.7 mm)(美国Thermo Fisher Scientific公司);Phoenix WinNonlin 7.0数据处理软件(美国Pharsight公司);XPE105电子天平(瑞士Mettler Toledo公司);ST16、ST16R离心机,ULTS1368医用低温保存箱(美国Thermo Fisher Scientific公司)。
1.3 方法
1.3.1 临床试验方案
根据文献报道,在一项奥美拉唑肠溶胶囊空腹生物等效性研究中,奥美拉唑Cmax个体内变异系数(CV%)为26.80%[4]。
参考《药物制剂人体生物利用度和生物等效性试验指导原则》(2015年版),考虑到脱落率、制剂间的差异以及拉唑类药物代谢存在基因多态性,故最终确定空腹生物等效性试验采用两周期、双交叉试验设计,纳入36例受试者。
由于奥美拉唑肠溶胶囊的崩解与释放受食物与生产工艺的影响较大,故餐后生物等效性试验采用三周期、部分重复交叉试验设计,纳入48例受试者。
本研究严格遵循“药物临床试验质量管理规范(GCP)”,并且已得到武汉市第四医院伦理委员会审查批准(临床试验登记号:CTR20201363)。所有受试者均通过体格检查,并自愿签署知情同意书。
1.3.2 给药方法与血样采集时间点设计
1)空腹组 受试者需在前一晚晚餐后禁食超过10 h。次日早8:00餐前口服受试或参比制剂(20 mg/人),用240 mL温水送服。服药前后1 h禁水,服药后4 h禁食。采血时间点包括服药前1 h至服药后24 h共24个时间点。
2)餐后组 受试者同样需在前一晚晚餐后禁食超过10 h。次日早餐后30 min口服受试或参比制剂,服药前后1 h禁水(除早餐及服药用水),服药后4 h禁食。采血时间点包括早餐前至服药后24 h共24个时间点。给药方案见表1。

1.3.3 血浆样本处理
采集的血液样品置于EDTA-K2抗凝的真空采血管中并轻轻颠倒6次与抗凝剂混匀,室温暂存;离心(4 ℃、1 700 g、10 min),待升温至室温后取约700 mL上层血浆于检测管中,剩余上层血浆置于备份管中。样本从采集到分离血浆后置于?20 ℃冰箱暂存,并于24 h内转移至?80 ℃冰箱保存。生物样本的预处理及保存均在避光条件下进行。
1.3.4 药物浓度检测
采用经方法学验证的液相色谱-串联质谱法(LC-MS/ MS)测定受试者服药后血浆中奥美拉唑的血药浓度。采用梯度洗脱程序,流速0.8 mL/min,流动相A(0.1%甲酸水溶液)与B(0.1%甲酸乙腈溶液)的初始比例为99∶1,该流动相比例在20 min的分析时间内会呈线性梯度转变至A-B=1∶99[5-7]。
1.4 统计方法
采用Phoenix WinNonlin软件以非房室模型计算药代动力学参数。对于浓度低于定量下限的样品,在进行药代动力学分析时,在达到Cmax以前取样的样品应以“0”计算,在达到Cmax以后取样的样品应以“ND”计算。按照给药分组,采用样本量、算术平均值、标准差、变异系数、中位数值、四分位数、最小值、最大值以及几何平均值汇总显示主要药代动力学参数分析结果。药代动力学参数数据来源于临床试验统计分析报告。
对药代动力学参数Cmax、AUC0~t、AUC0~∞进行统计分析,显著性水平为0.05。AUC和Cmax需进行对数转换后进行随机效应模型的方差分析。随机效应模型的方差分析将总变异分解为顺序变异、周期变异、处理变异(受试制剂与参比制剂所引起的变异)、个体间的变异和个体内的变异(残差)。
采用mixed linear model置信区间法进行生物等效性评价。受试制剂的药代动力学参数Cmax、AUC0~t、AUC0~∞的90%置信区间为参比制剂相应参数的80.00%~125.00%范围内,可判定两制剂具有生物等效性。
2 结果
2.1 血药浓度检测
血浆中奥美拉唑分析测定方法的回归方程为y= 0.016 5x+0.005 69(R2=0.998 0),线性范围为1.500~1 500 ng/mL,定量下限(LLOQ)为1.500 ng/mL,质控样品数至少为未知样品数的5%,全部质控样品的合格率均符合要求。
QC样品浓度分别为4.500、45.00、450.0、1 125 ng/mL 4个浓度水平奥美拉唑每一浓度水平QC样品的精密度(CV%)均不大于9.51%,准确度偏差范围为?0.33%~0.52%。
2.2 空腹给药试验药代动力学
35例受试者完成两周期服药及采血(单剂量20 mg),主要药代动力学参数对比结果见表2,药时曲线结果见图1。结果表明,空腹口服受试制剂及参比制剂后,两者的Cmax、AUC0~t、AUC0~∞的几何均值比值(T/R)的90%置信区间均在80.00%~125.00%范围内,符合生物等效性接受标准,具有生物等效性。

2.3 空腹给药试验安全性
发生的不良反应的例数为7例,发生率为19.4%(7/36),T药例数5例、发生率为14.3%(5/35),R药例数3例、发生率为8.3%(3/36)(同一受试者可能发生不同类别的不良反应以及对不同的药物发生不同的不良反应)。不良反应系统分析见表3。

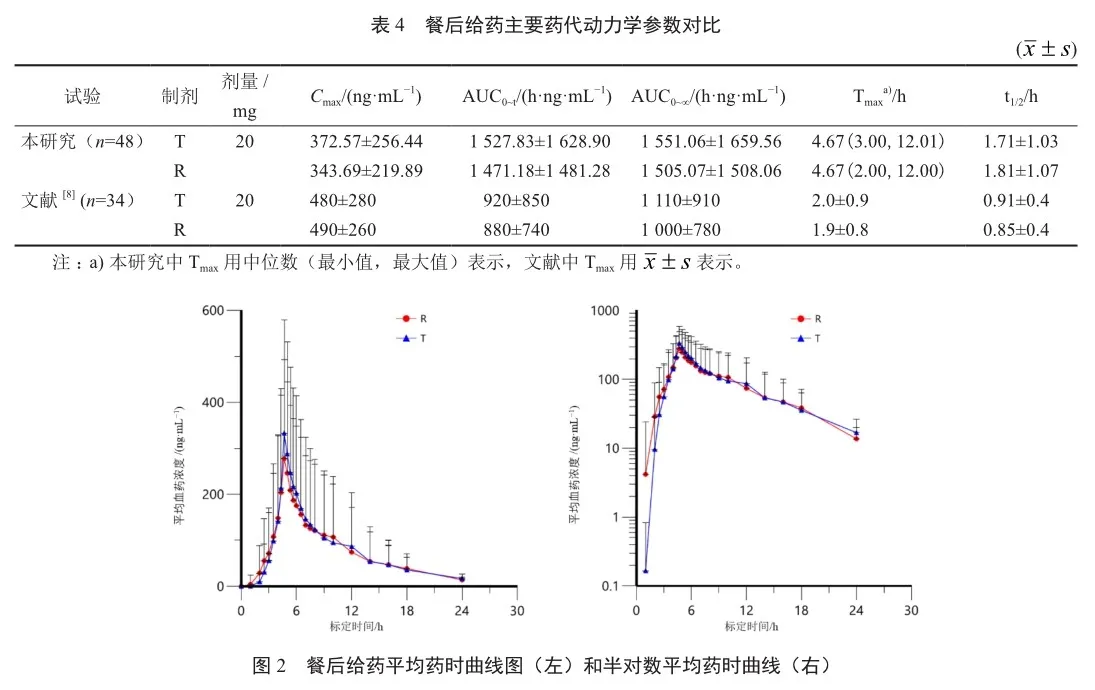
2.4 餐后给药试验药代动力学
48例受试者完成三周期服药及采血餐后给药试验,主要药代动力学参数对比结果见表4,药时曲线结果见图2。
结果表明,奥美拉唑肠溶胶囊的药代参数Cmax的参比制剂SWR为0.405,SWR≥0.294,则采用RSAB进行评价,Cmax的几何均数比值(受试制剂/参比制剂)的点估计值及单侧95%置信区间上限,等效界值点估计值为1.005 1不超过80.00%~125.00%,单侧95%置信区间上限?0.0927≤0,符合生物等效标准;AUC0~t、AUC0~∞的参比制剂SWR均为SWR<0.294,则采用平均生物等效性的统计方法进行评价,受试制剂与参比制剂的药代动力学参数AUC0~t、AUC0~∞几何均值比值的90%置信区间分别为93.21%~106.81%和92.39%~105.09%,均在80.00%~125.00%范围内,符合生物等效性的接受标准,即受试制剂和参比制剂在餐后给药条件下具有生物等效性。
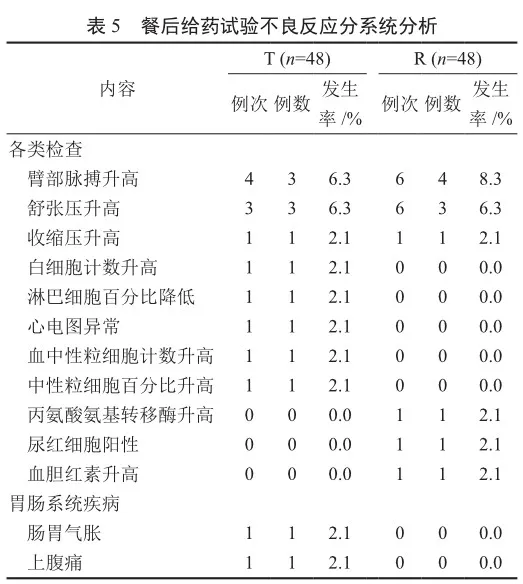
2.5 餐后给药试验安全性
发生的不良反应的例数为10例,发生率为20.8%(10/48),R药例数7例、发生率为14.6%(7/48),T药例数6例、发生率为12.5%(6/48)(同一受试者可能发生不同类别的不良反应及对不同的药物发生不良反应)。不良反应系统分析见表5。
3 讨论
根据2016年原国家食品药品监督管理总局发布的《以药动学参数为终点评价指标的化学药物仿制药人体生物等效性研究技术指导原则》以及美国食品药品管理局颁布的《奥美拉唑肠溶胶囊生物等效性指导原则》进行了临床研究,对受试制剂和参比制剂的主要药动学参数,包括Cmax、AUC0~t、AUC0~∞进行了对比分析,结果显示两者几何均值比的90%置信区间均符合生物等效性评价标准,生物等效性结果具有高度的可靠性。对比文献的临床试验结果,充分体现了不同工艺方法制备的奥美拉唑肠溶胶囊在体内吸收行为具有显著差异,验证了奥美拉唑体内吸收的高变异性。此外,受试制剂与参比制剂在不良事件发生率上相近,且所有不良反应均为轻度,安全性无显著差异。本研究纳入受试者均为18~65岁的健康人群,未涉及老年、低龄和特殊人群的样本研究,研究结果的普遍性可能受到一定限制,仿制药品上市后仍需关注真实世界数据及药物警戒信息。
本研究所用的两种制剂在空腹和餐后两种条件下均展现出良好的生物等效性和安全性,故在临床应用中可相互替代。
参考文献
[1] 张可翔. 奥美拉唑肠溶胶囊一致性评价产品处方工艺改进探讨[J]. 中国科技期刊数据库-医药, 2021(9): 172-174.
[2] 袁鹰, 肖帆, 常晓敏. 自制奥美拉唑肠溶胶囊与参比制剂的体外溶出行为一致性评价[J]. 化学与黏合, 2019, 41(2): 117-120.
[3] 马婧怡, 贺锐锐, 王骏. 肠溶制剂餐后生物等效性试验中的考虑[J]. 中国临床药理学杂志, 2022, 38(18): 2244-2248.
[4] 刘曼, 刘会臣. 对国内奥美拉唑肠溶制剂生物等效性试验的分析[J]. 中国新药杂志, 2023, 32(6): 605-609.
[5] U.S. Food and Drug Administration. Bioanalytical method validation guidance for industry[EB/OL]. (2018-05-24)[2024-04-08]. https://www.fda.gov/media/70858/download.
[6] European Medicines Agency. Guideline on bioanalytical method validation[EB/OL]. (2011-07-21)[2024-04-08]. https://www.ema.europa.eu/en/documents/scientificguideline/guideline-bioanalytical-method-validation_en.pdf.
[7] Kumisbek G, Vetchy D, Kadyrbay A. Development of a new bioequivalent omeprazole product[J]. Medicina (Kaunas), 2024, 60(3): 427.
[8] Poo JL, Galán JF, Rosete A, et al. Bioavailability of two single-dose oral formulations of omeprazole 20 mg: an openlabel, randomized sequence, two-period crossover comparison in healthy Mexican adult volunteers[J]. Clin Ther, 2008, 30(4): 693-699.
