
锂硫电池中的硫正极电催化认识
2024-07-04 00:00:00汪涛董琴李存璞魏子栋
物理化学学报 2024年2期
关键词:电催化


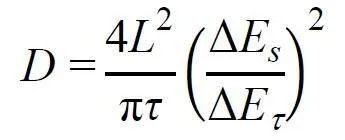


摘要:以单质硫为正极的锂硫电池表现出极高的放电比容量(1672 mAh·g−1),是极具潜力的下一代二次动力电池。然而,充放电过程中溶解的高阶多硫化锂(Li2Sn,4 ≤ n ≤ 8)的穿梭效应,以及硫物种缓慢的氧化还原动力学过程是锂硫电池商业应用前需要解决的关键问题。而电化学催化的引入是解决上述问题行之有效的策略。本文从电化学催化角度出发,重新讨论认识多硫化物的存在形式,并从吸附-催化、活性中间体两个方面,根据不同的反应机理、路径分析多硫化物转化机制,总结定量评价催化性能方法,以期为锂硫电池高效电催化剂的设计提供思路。
关键词:锂硫电池;催化转化;电催化;化学吸附;硫自由基
中图分类号:O643
1 引言
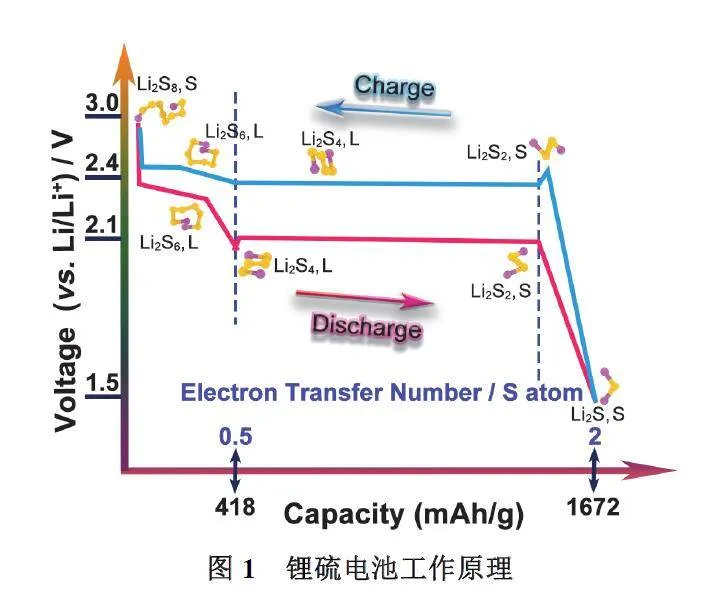
基于超高的能量密度、高安全性以及成本低廉等优势,锂硫电池是最具发展潜力的新型储能器件之一1–12。典型的锂硫电池主要是由锂金属负极,有机电解液,隔膜和硫正极材料构成。硫单质在自然界中主要以稳定的皇冠型S8分子存在。而锂硫电池的放电过程正是S8分子中的S―S键不断发生电化学断裂以及重新键合的过程(图1),当正极中的S8分子与锂离子完全反应时,释放出大量电能,其电化学反应方程式如下:
16Li+ + S8 + 16e− → 8Li2S
然而,由于硫最外层有6个电子,通过裂分产生不同状态硫物种,因此硫在正极进行的是多步骤、多电子、多相的电化学反应。普遍认为,硫在放电过程中存在两个相变过程(固液转化I,液固转化II)以及两个放电平台(2.4和2.1 V)。电池放电的过程中,负极的金属锂失去电子变成锂离子,电子通过外部电路转移到正极,此时正极上的单质硫得到电子在电解液中与锂离子结合形成可溶性高阶多硫化物(S8 → Li2S8 → Li2S6 → Li2S4;固液转化I),对应着在2.4 V左右出现第一个平台,理论上每个硫原子接纳0.5个电子,释放出418 mAh·g−1的比容量,对应的电化学方程式如下:
S8 + 4Li+ + 4e− → 2Li2S4
随后可溶性高阶多硫化物继续得到电子分解成不可溶性的低阶多硫化物(Li2S4 → Li2S2 →Li2S;液固转化II),最后沉积到正极上,对应着在2.1 V左右出现第二个平台。作为长放电平台,该过程承担了电池的主要放电容量,对应的电化学方程式如下:
Li2S4 + 4Li+ + 4e−→" 2Li2S + Li2S2
虽然锂硫电池在二次电池体系中优势突出,但是还有一些关键性问题需要解决,其中因穿梭效应带来电池容量的快速衰减以及低循环寿命严重制约了锂硫电池商业化进程13–21。所谓穿梭效应,是指在锂硫电池放电过程中,多硫化物不断生成并溶解于电解液中,造成活性物质与电极表面的脱离并进一步导致放电产物的大量集聚,降低整个电池的反应速率,并且由于浓度差自发迁移到负极,在负极区域形成不可逆转的“死硫”;而在充电过程中,不溶性放电产物具有较差的导电性,大量堆积造成电池内阻急剧增大,同时固态放电产物向可溶性多硫化物的转化需要较高的活化能,也会降低电池的反应速率和电池的能量效率22–32。
如何促进多硫化物快速转化成为解决穿梭效应的关键性因素,近年来,催化效应逐渐被引入到穿梭效应的解决方案中,其作用原理犹如在人体内注入“活性酶”来实现有毒废物的高效转化一样,催化材料能够“主动”加速多硫化物的快速可逆转化,降低多硫化物与电解液的接触概率,进而提高电池效率33–44。
2 对多硫化物存在形式的认识
尽管在调节多硫化物的溶解度和溶剂化结构方面已经取得了显著进展,但多硫化物在电解液中的实际存在形式尚未明确揭示,特别是Li+和多硫化物阴离子(Sn2−,n = 4、6或8)之间的解离和缔合行为。换言之,溶解的Li2Sn分子是否以及如何被电离以及其与周围的电解质之间作用关系仍然存在争议。1973年Sawyer等45通过循环伏安法、控制电位电解法和吸收光谱法研究了二甲基亚砜(Dimethyl sulfoxide,DMSO)溶剂中硫的还原过程:S8得到电子还原成S62−离子,进一步得到电子还原成S42−离子。基于此,传统观点认为多硫化锂类似常见的锂盐在电解液中充分解离,主要以多硫阴离子(S n2−)形式存在。
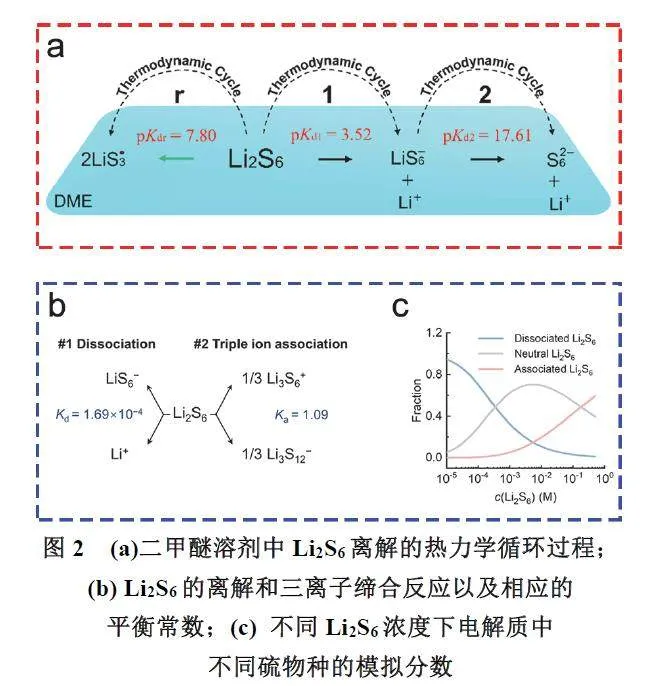
随着研究的深入以及检测技术的快速发展,近年来, Rajput 等46 通过分子动力学(MolecularDynamics,MD)模拟研究了锂硫电池电解液中电解质添加剂和多硫化物之间的相互作用,提出了多硫化物倾向于与强Li+-S n2−相互作用聚合。Gao等47利用从头算方法计算了Li2S6在1,2-二甲氧基乙烷(1,2-Dimethoxyethane,DME)中的解离平衡常数(图2a)。研究发现,Li2S6在DME中主要以中性分子形式存在,多硫化物阴离子和多硫自由基则较为少见。
Zhang等48发现锂硫电池中除了多硫化物分子存在外,还存在多硫化锂阳离子(Li3S6+),通过电喷雾离子质谱(Electrospray Ionization MassSpectrometry,ESI-MS)测试,在Li2S6溶液中加入额外的锂盐(LiTFSI)后,质谱中观察到了显著而明确的多硫化锂阳离子Li3S6+的特征峰。此外,变温7LiNMR t1弛豫时间测试表明,加入的额外Li+倾向于与Li2S6结合,从而表现出相应的特征配位环境。紫外-可见光谱进一步表明,这种相互作用明显改变了多硫化物的电子结构。这些实验证明了在锂盐为添加剂的电解液中,Li2S6分子具有缔合游离Li+生成Li3S6+阳离子的倾向(图2b,c)。
综上所述,我们对锂硫电池中多硫化物的存在形式有了明确的认识, 在常规电解液溶剂(DOL/DME)中多硫化物主要以中性分子Li2Sn存在,一小部分会发生电离形成S n2−阴离子,一小部分会倾向于与Li+结合形成阳离子Li3S6+。基于此,针对多硫化物不同的存在形式可以设计不同作用机理的电催化剂。
如果是针对多硫化物中性分子,则各种宿主材料可以通过亲锂主体和亲硫主体或两者的协同作用诱导吸附中性Li2Sn分子,通过界面吸附-催化策略促进多硫化物的快速转化。例如多孔碳材料、金属化合物,含有极性键的有机化合物等42,49,50。
相反,如果是针对多硫化物阴离子,则硫正极处的正电位点将更适合于阻止多硫化物的穿梭。而由于额外的电迁移,溶解的多硫化物阴离子可能比仅由浓度差驱动的中性Li2Sn引起更严重的穿梭效应。催化多硫化物阴离子形成活性中间体加速其转化可有效抑制穿梭效应,例如形成硫自由基,硫酸盐51,52。多硫化物在DOL/DME溶剂中的存在形式:
最后,如果针对多硫化物阳离子,低浓度锂盐可以当作添加剂通过共离子效应抑制多硫化物的溶解48,53。
3 催化策略——基于不同的电化学反应路径选择
催化的概念最早由瑞典的贝采里乌斯(JonsJakob Berzelius)提出,他指出物质间除了静电作用引起化学力之外,还有一种力,“Catalytic Force”,即催化力。这种作用可以把物质拆分成元素随后重新整合,但物质本身不变。他把这种作用称为“catalysis”,即催化作用。催化作用是一种能够改变反应速率而不影响反应平衡的物理化学作用,它不是单纯的物理吸附作用,同时虽然参与了反应,改变了反应途径,降低了活化能,但不会影响产物和产率。而锂硫电池中硫还原反应(SulfurReduction Reactions,SRRs)和硫氧化反应(SulfurOxidation Reactions,SORs)动力学缓慢过程引起的穿梭效应是导致锂硫电池暂时不能商业化应用的重要因素。因此将电催化剂引入锂硫电池体系催化多硫化物的快速转化,成为目前抑制穿梭效应的高效策略。然而传统的氧化还原催化主要是通过调控吸附状态,促进化学键的断裂耦合。锂硫电池中由于硫的氧化还原是多步且非均相反应,因此,锂硫电池中的催化作用主要是指促进不同硫物种的快速转化、形成额外的反应路径达到“弯道超车”的效果以及形成活性中间体调控决速步骤5,54–58。
3.1 “吸附-催化”策略
吸附是一种固体表面现象。由于固体表面分子所处的位置不同,则固体分子之间的力不可能处于平衡。因此,固体吸附剂表面具有一种吸引力,吸引着接触它的分子,这便是所谓的吸附作用。无论是长链还是短链多硫化物都由于末端硫带负电荷而表现出相同的极性,理论上极性材料的表面都可以提供与多硫化物之间的强亲和力,当两者之间仅有分子间作用力,也就是范德华力时,此时就处于物理吸附的状态,而当两者之间有新的化学键的耦合以及断裂时,可称之为化学吸附。化学吸附是吸附质分子与固体表面原子(或分子)发生电子的转移、交换或共有,形成吸附化学键的吸附。而物理吸附仅仅是能量的调控,并不涉及轨道效应。锂硫电池体系中,无论是物理吸附还是化学吸附,都是将多硫化物聚集在吸附材料周围,并建立起不同程度的相互作用力。这种相互作用能将增强电荷的转移能力,并催化多硫化物的转化。与单纯的碳质材料相比,它可以实现吸附和催化的协同作用,从而大大提高化学性能。
3.1.1 物理吸附-能量的调控
物理吸附从本质上来说就是对多硫化物和材料表面之间的一个能量的调控过程。当两者之间存在分子间作用力,基底材料将多硫化物吸附到周围,使其产生聚集,这时基底材料的高导电性会加速电子的转移,从而促进电池内部氧化还原反应的进行,也就是我们常说的催化多硫化物的迅速转化,达到抑制穿梭效应的效果11,21,38。
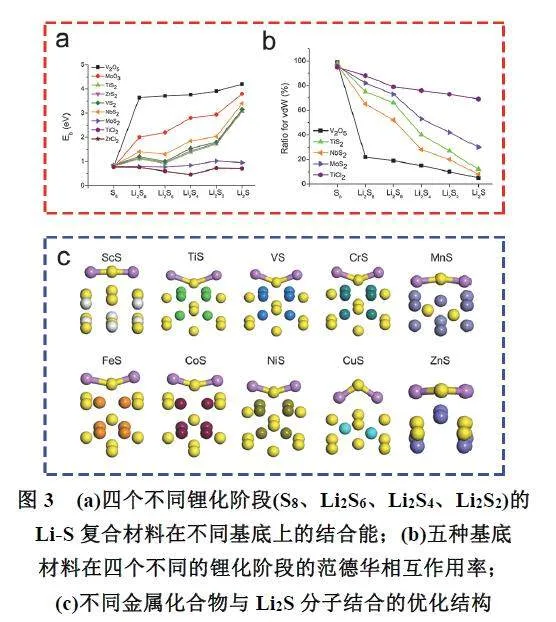
Cui等59使用范德华相互作用的第一性原理方法,系统地研究了Li2Sn物种在各种二维层状材料(氧化物、硫化物和氯化物)以及石墨烯材料上的吸附(图3a,b),并详细的研究了两者之间的相互作用力大小和电子分布情况,其中包括结合强度、构型畸变和电荷转移量。研究发现:不同的材料与多硫化物具有不同的吸附特征,而吸附强度由多硫化物簇中S原子向吸附材料的电荷转移量决定。而这种物理锚定总是伴随着多硫化物中Li―S键的软化,当吸附能过低时多硫化物无法被很好地固定在材料表面;而吸附能过高时,过强的结合反而会导致Li2Sn物种的破坏,加速多硫化物的溶解扩散。因此对多硫化物的吸附能应该控制在一个合适的范围内(0.8–2.0 eV)。Zhang等60基于离散傅里叶变换(Discrete Fourier Transform,DFT)计算深入分析多硫化物吸附于不同金属硫化合物表面的电子结构,解析了不同结合能的相互作用机制和起源(图3c)。研究发现多硫化物与金属硫化合物之间的强相互作用通常会有一个显著的电荷转移,这主要源于两者的d–p轨道相互作用。Zu等61通过简单的生物模板法合成了各种非导电金属氧化物纳米颗粒修饰的碳片(氧化镁、氧化铈、氧化镧、氧化铝、氧化钙),DFT计算和静态吸附实验证实了单层吸附在Li2Sn捕获过程中占主导地位。此外还发现,这些金属氧化物,特别是一些非导电氧化物充当了Li2Sn中转站的角色,将Li2Sn从导电性差的氧化物表面输送到导电性高的碳材料基底上,以确保快速的电化学转化。Guo等43基于系统-环境理论,从热力学角度出发,重新审视了非插层电极材料在空间限域体系中的电化学反应过程;概述了硫元素正极材料限域结构设计的最新进展;述评了空间限域效应对正极材料的电荷转移动力学、结构演变、以及电化学反应机制的重要影响;讨论了结构与性能的构效关系。
因此,这里我们将锂硫电池中的物理吸附-催化定义为基底材料与多硫化物之间存在较弱分子间作用力,能将多硫化物吸附在材料表面促进其转化,尽管这种作用力带来的效果是有限的。传统的导电多孔碳材料(介孔、微孔、分级多孔,石墨烯,碳纳米管)便是通过物理吸附-催化策略来解决硫导电性差问题的同时有效抑制穿梭效应61–63。
3.1.2 化学吸附-轨道效应
化学吸附本质上就是吸附物种和基底材料之间的轨道效应。就锂硫电池体系而言,“化学吸附-催化”指的是基底材料与多硫化物在界面上发生化学键的耦合断裂来促进多硫化物转化,即化学反应调控电化学反应过程。针对多硫化物的存在形式,锂硫电池中的化学吸附可以分为两类:一是掺杂电负性高的非金属元素X (N、O、P、S、B等),这类元素易与多硫化物中的Li原子发生电荷转移,形成“Li-X”,从而产生对多硫化物的强化学吸附作用力64–66;二是利用过渡金属元素M (Ti、V、Cr、Mn、Fe、Co、Ni、Mo等)电子能级较低,3d轨道上的电子数较少特点,更容易失去电子与多硫化物中的S原子耦合,形成新的化学键“M―S”键,将多硫化物牢牢限制在材料表面67,68。
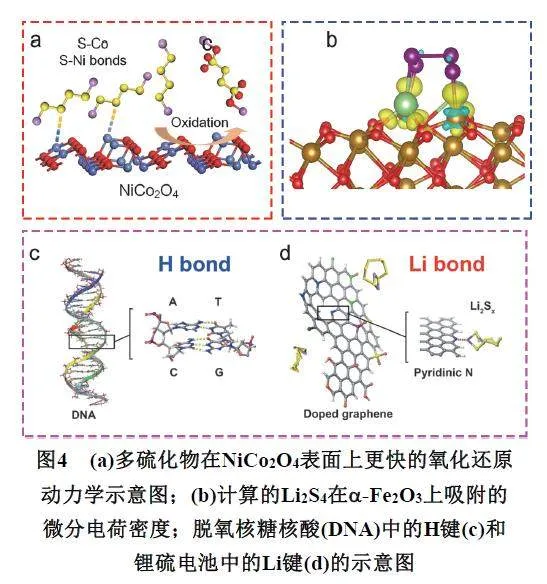
Gao等69报道了一种硫载体S/NiCo2O4,通过Ni和Co分别和多硫化物种的S原子形成“Ni―S”、“Co―S”达到吸附可溶性多硫化物的效果(图4a)。Yang等70通过一步水热处理,在3D分级多孔石墨烯上制备了均匀分布的α-Fe2O3纳米颗粒,通过计算分析了Fe2O3-Li2S4体系的差分电荷密度以及相应的XPS数据,证明了α-Fe2O3-Li2S4体系中存在“Fe―S”键以及“Li―O”键(图4b),进一步分析发现,α-Fe2O3与多硫化物之间的电荷转移量决定了两者之间的吸附能大小。Liu等71设计合成了具有不对称配位构型的Fe单原子催化剂(Fe-N3C2-C),实验和理论结果表明,非对称配位的Fe-N3C2-C不仅通过加强sp轨道与Fe dx2−y2/dxy的d-p轨道杂化强调以形成额外π键来增强对多硫化物的锚定能力,而且还通过降低Li2S沉淀/分解能垒来增强多硫化物氧化还原动力学,从而抑制穿梭效应。Zhang等72,73通过模拟不同边缘结构的氮掺杂石墨烯纳米带(GNRs)与多硫化物之间的吸附行为,提出对导电骨架进行化学修饰,可有效增强多硫化物与硫宿主之间相互作用,从而建立“强耦合界面”,维持稳定有效的电化学接触。此外,从“锂键”化学的角度出发(图4c–d),在原子水平上阐释了多硫化物与吸附位点之间强路易斯酸碱相互作用的本质起源,并确立了多硫化物与吸附位点间相互作用的关键描述符,即偶极–偶极相互作用的强度。通过构建描述符衡量不同宿主材料的吸附能力,解决实验在定量描述宿主材料吸附性能方面的困难。
因此,我们将锂硫电池中的化学吸附定义为基底材料与多硫化物之间存在明显的轨道作用。例如,非金属元素掺杂、单原子掺杂、金属化合物等通过与多硫化物中的Li或者S原子产生化学键的耦合/断裂,从而促进多硫化物的快速转化来达到化学吸附-催化的效果74–79。
总的来说,功能性复合正极材料的设计不应局限于单一的物理吸附或者化学吸附,而是将两者结合起来,通过各自不同的作用机制共同催化多硫化物的快速转化,达到吸附-催化的效果。
3.2 活性中间体策略
锂硫电池内的反应极其复杂,充放电过程中元素硫要经历一系列氧化以及还原过程。虽然Li2S是最终还原产物,Li2S8是最终氧化产物,但通过引入或者改变活性中间体的状态以及形式可以得到不同的反应路径,调控不同的决速步骤,从而产生不同的催化效果。
3.2.1 硫自由基活性中间体
自由基,化学中定义为共价键发生均裂而形成的具有不成对电子的原子或基团。首先它是真实存在的电中性中间体,其次,由于其含有不成对电子,导致化学性质非常活泼,极易发生氧化还原反应。在锂硫电池体系中,已被报道的常见自由基有S3 · 以及S2 · 。
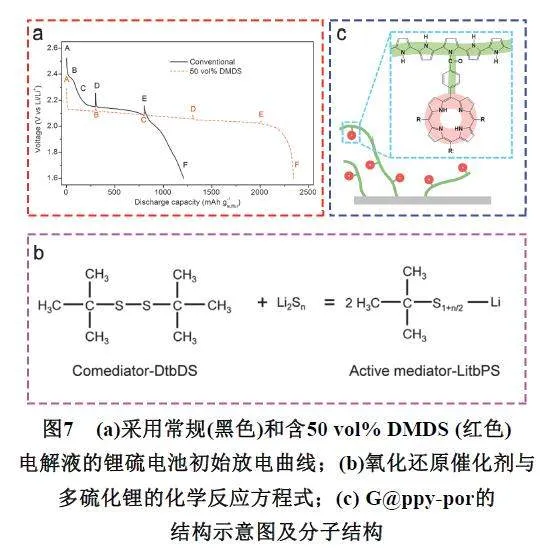
Alloin等80通过液相色谱、紫外-可见吸收和电子自旋共振光谱,研究了在四乙二醇二甲醚基电解液中,不同电位下电解质的组成。研究发现,在S62−转化为S42−的过程中由于歧化反应会产生S3 ·−中间态。Wei 等81 报道了一种含有硫空位的Co9S8/MoS2的异质结,硫空位的引入诱导形成S3 · ,提供一条新的反应路径,加速了多硫化物转化及成核(图5a)。当存在硫空位时,Li2S6的一个硫原子会倾向于填充至硫空位附近,以降低整个吸附体系的能量。同时Li2S6将转化为活泼的Li2S5,Li2S5进一步转化为2个LiS3 · 自由基。随后活泼的LiS3 · 在电极界面获得电子转化为Li2S3,高活性的Li2S3进一步的与Li2S4在电解液中发生歧化反应,生成最终放电产物固态的Li2S与Li2S6,Li2S6重新进入新的一轮循环,而Li2S则成为后续的成核位点。Nazar等82发现S3 ·−可以稳定存在于电子对供体(ElectronPair Donor,EPD)电解液中,并以二甲基乙酰胺(Dimethylacetamide,DMAC)作为溶剂进行了充放电机理研究。EPD由于具有较高的供体数量,能促进多硫化物的解离,从而形成S3 ·−。此外EPD与S3 ·−的相互依存也能加强多硫化物之间的电化学平衡。在放电过程中,直至含硫物质完全还原前,S3 ·−可以重新氧化一小部分Li2S,防止正极持续钝化。在充电过程中,EPD可以部分溶剂化固体Li2S,缓解固体Li2S导电性差,动力学过程缓慢。Wei等83提出了“吸的强不一定好,转化快才是真的强”策略,合成了一种生长在碳布上的1T MoS2-MnO2异质结构作为锂硫电池的电接触覆盖层。DFT计算发现,单独的MoS2对多硫化物的最强吸附集中在Li2S4,这种强吸附力虽然能将Li2S4聚集在材料表面,但却不利于进一步转化为固体Li2S。然而当无定形MnO2引入与MoS2结合形成异质结,发现整个体系的最强吸附力集中到了LiS2 · 。因此通过这种调节多硫化物的整体催化转化行为,将多硫化物级联转化反应的决速步骤(Speed DeterminationSteps,SDS)液体-固体过程(Li2S4 → LiS2 · )改变为固体-固体过程(吸附界面LiS2 · → Li2S2),溶解的多硫化物还来不及转移到负极侧就在覆盖层上就地转化为固体LiS2 · ,然后实现LiS2 · 到Li2S2/Li2S的均匀成核。因此多硫化物可以随时从正极分解出来,但永远不会从覆盖层基底离开(图5b)。
锂硫电池体系中,处于中间态的多硫化物会发生歧化反应,这是导致产生硫自由基的直接原因。自由基在电催化剂上的吸附吉布斯自由能决定了转化过程的反应机制、决速步骤及所需过电势等关键信息。而杂原子的掺杂、异质结、缺陷位、空位的引入等都易诱导硫自由基的产生52,84,85。
3.2.2 硫代硫酸盐或聚硫酸盐活性中间体
多硫化物本身就容易发生级联反应,一些金属氧化物可以一部分将多硫化物转化为硫代硫酸盐(S2O32−),长链Sn2− (n gt; 4)与可以与S2O32−中的S―S键相连,生成聚硫醚[S2O32-(S)x−3-S2O3]2−。这些聚硫醚中链状S―S键具有强电化学活性,可以吸附锂离子,增强氧化还原反应动力学。
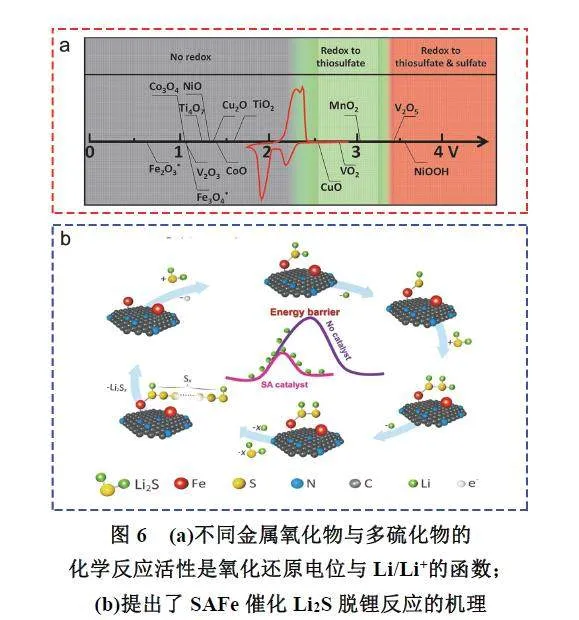
Nazar等86研究发现S2O32−的形成与过渡金属氧化物氧化还原电位相关(图6a),氧化还原电位低于1.5 V的金属氧化物(如Co3O4或Ti4O7)与多硫化物之间尽管存在强烈的极性-极性和路易斯酸碱相互作用,但并没有出现氧化反应形成S2O32−。当氧化还原电位处于1.5–3.05 V时,金属氧化物(如MnO2)能有效吸附多硫化物并将其氧化成活性中间体S2O32−,当氧化还原电位大于3.05 V时,金属氧化物(如V2O5)将多硫化物反复氧化为无电化学活性的硫酸盐基团(SO42−),造成正极材料不可逆损失。Sun等87报道了一种生长在氮掺杂的碳纳米管(NCNT)阵列上的VO2纳米颗粒作为硫载体材料,VO2纳米颗粒起到催化剂的作用,氧化多硫化物以产生活性中间体硫代硫酸盐,硫代硫酸盐作为介体将长链多硫化物耦合在一起,并将其快速转化为短链Li2S2/Li2S。Yang等88发现将In2O3基催化剂引入正极材料能有效提高锂硫电池电化学性能,但在循环一段时间后In2O3催化剂会因表面“中毒”而失活。原位拉曼光谱实时监测发现,在完全放电状态下,In2O3基正极材料的硫酸盐含量从第一次循环的31.6%增加到64.7% (1000次循环后),因此提出不可逆硫酸盐的大量累积是造成催化剂失效的重要原因。当然,除了金属氧化物外,其他材料也能将多硫化物氧化成硫酸盐。Zhang等89采用原位和非原位X射线吸收光谱、电化学分析,以及理论模拟等方法揭示了铁单原子催化剂(FeSingle Atomic Catalyst,SAFe)在Li2S在脱锂中的作用,结果发现铁单原子可以通过原子表面与硫原子之间的强亲核作用,在促进相对惰性Li2S在还原的过程中,形成长链聚硫酸盐活性中间体,加速反应动力学过程,组装的锂硫电池实现了在超高倍率(12C)下稳定循环(图6b)。
因此,硫代硫酸盐或聚硫酸盐的形成是由于基底材料的氧化作用,主要是具有强氧化性的金属化合物。他们在材料表面将多硫化物氧化为不溶的硫代硫酸盐,随后形成聚硫酸盐,并快速通过内部歧化反应转化为固体Li2S2或者Li2S。然而不可逆硫酸盐的大量累积也不是一件好事,可能会导致催化剂“中毒”从而失效88,90,91。
3.2.3 有机硫分子中间体
在锂硫电池体系中直接引入有机含硫分子作为正极硫供体,或者通过形成有机硫分子活性中间体来加速氧化还原反应的进行,也是抑制穿梭效应行之有效的方法。
Wang等92报道了一种含有二甲基二硫化物(Dimethyl Disulfide,DMDS)作为共溶剂的功能性电解质系统,发现DMDS与硫反应形成可溶性甲基封端多硫化物中间体,在放电过程中进一步还原为有机硫化锂,这意味着DMDS中的硫元素作为硫源参与了放电过程,提供了额外的反应路径,基于这种新的反应机制,含有DMDS的电解质为电池提供了额外的容量,几乎达到了传统锂硫电解质电池的两倍(图7a)。Hunag等93由生物学中的辅酶的概念受到启发,提出了锂硫电池氧化还原辅助介体(Redox comediator,coRM)的概念,将有机多硫化物( 二叔丁基二硫化物, tert-Butyldisulfide,DtbDS)介体加入电解液中,它能与多硫化物发生化学吸附,生成一端为叔丁基、一端为(CH3)3C-Sx-Li多硫化物作为活性中间体参与反应(图7b),与硫直接还原得到的多硫化物相比,(CH3)3C-Sx-Li活性中间体具有更快的氧化还原动力学过程,可加速Li2S的成核和分解。Seo等94首次提出了利用有机小分子蒽醌(Anthraquinone,AQ)“固硫”的创新思路,实现了高载量硫正极长期循环的稳定性。作者研究发现蒽醌小分子可迅速与多硫化物作用形成蒽醌沉淀物(AQ-Li2S4),这种作用不是简单的复合,而是发生了可逆的氧化还原反应形成S―O化学键,从而使蒽醌小分子对可溶性多硫化物产生较强的吸附作用。据此,研究者提出了不同于传统的有机小分子助力的“固硫”新机制,即通过在充放电过程中小分子蒽醌与可溶性多硫化锂发生“化学性吸附”,形成无法溶解于电解液的不溶性产物,从而实现了对活性物质流失的有效抑制,显著地增加了电池的寿命。Zhang等95设计了一种半固定化的分子电催化剂,以促进工作中的锂硫电池的多相硫氧化反应的动力学。具体来说,将卟啉活性位点共价接枝在导电且柔性的高分子链上,一方面使得活性位点高效“捕获”液相中间物种多硫化物以行使均相催化功能,另一方面也使得活性位点有效与电子通路相连从而行使异相催化功能(图7c)。
近年来,有机分子由于其丰富的官能团结构受到越来越多锂硫电池研究者的青睐。然而其本身导电性差的特性以及活性硫物质含量低的问题是目前亟待解决的难题96–99。
4 定量评价催化性能的测试方法
基底材料催化多硫化物转化性能的强弱需要定量的指标进行评价,以建立规范的评价标准。因此,本节主要简单介绍目前评价锂硫电池催化性能的常规测试方法。
4.1 循环伏安曲线
循环伏安法(Cyclic Voltammogram,CV)是一种研究电极/电解液界面上电化学反应行为的技术手段,可以得到电化学反应机理(峰信息)、反应速度(塔菲尔斜率)和电极过程动力学参数(成核转化因子)等。
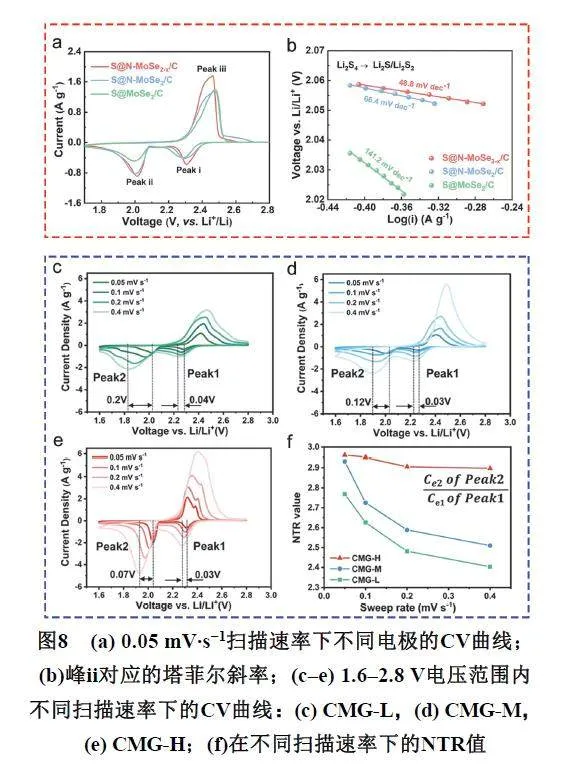
典型的锂硫电池循环伏安曲线中均存在两个还原峰,负扫的第一个还原峰在2.3 V附近,对应的是单质硫(S8)得到电子被还原为长链可溶多硫化物(Li2S8-Li2S4)的过程,第二个还原峰在2.0 V附近,对应的是长链可溶多硫化物(Li2S4)进一步得到电子转化为不溶的短链多硫化物(Li2S4-Li2S2/Li2S)的沉积过程(图8a) 5。可以通过对比分析材料的氧化还原特征峰来评估催化性能,比如氧化还原特征峰的偏移、出峰位置以及峰面积等。还可以通过拟合氧化还原峰得到塔菲尔斜率(TafelSlope),塔菲尔公式是塔菲尔于1905年提出的经验公式,证明了电极的极化反映了电极过程受阻的情况(图8b)。塔菲尔斜率的单位一般是mV·dec−1,表示的是电流变化十倍,过电位需要的变化量。塔菲尔斜率越小,电流升高10倍时所需电压的提升越小,对应能耗就越低。说明在相同动力学电流密度或表观电流密度下,该催化过程的过电势越低。因此,塔菲尔斜率也可以作为定量评价催化性能的参数。
最近,魏子栋课题组81从电化学角度出发,通过不同扫速下的CV数据,定义了成核转换因子(Nucleation Transformation Ratio,NTR)来定量评价材料催化性能(图8c–f):NTR = Ce2/Ce1其中Ce1和Ce2分别是与Peak1与Peak2对应的电量。在理想状态下,NTR = 3,某种材料的循环伏安测试所计算的NTR越大越接近于3,意味着从Li2S6到Li2S4的反应更加迅速,说明材料对多硫化物成核的催化作用越强,进一步的,随着扫速的增加,具有较强催化性能的材料可以始终提供足够的反应速率,NTR几乎不会变小;而催化性能较差的材料,由于循环伏安的电位会迅速掠过多硫化物成核反应的电化学窗口,Peak1的产物无法在Peak2反应快速转化,NTR会减小。因此成核转换因子NTR随CV扫描速度变化所展示的衰减趋势,可以帮助我们定量判断正极材料对硫及其多硫化物的吸附、限域和催化作用,使锂硫电池正极的研究从定性或笼统描述转入有具体因子的定量评价。
4.2 电化学阻抗谱
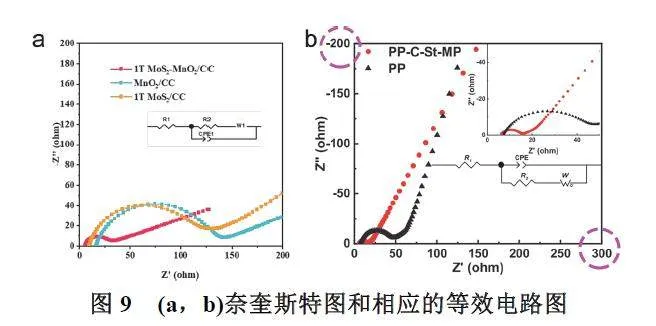
电化学阻抗谱(Electrochemical ImpedanceSpectroscopy,EIS)是指以小振幅的正弦波势电流为扰动信号,使电极系统产生近似线性关系的响应,测量电极系统在很宽频率范围内的阻抗谱图,以此来研究电池系统的方法。通过模拟由电阻、电感与电容串联、并联组成的等效电路图,结合电化学阻抗谱图,能够有效分析材料的导电性和离子扩散速度,从而评价多硫化物催化转化的速率。如图9所示83,化学阻抗谱图中高频区的半圆直径反映了电极材料内部的电荷转移阻力大小,拟合的半圆直径的值越小说明材料的电荷转移阻力越小,电子传输速率越快。而低频区的直线斜率反映了锂离子在电极材料内部的扩散阻力大小,直线斜率越大说明锂离子在电极材料内的扩散速度越快。值得注意的是,一些科研人员在处理Nyquist图谱时,横纵坐标并不一致,这会导致拟合半圆直径得到的阻抗数据并不准确(图9b) 100。
4.3 恒电流滴定法
恒电流滴定法(Galvanostatic IntermittentTitration Technique,GITT)可以定量分析Li+在电极材料内部的扩散速度。首先施加正电流脉冲,使电压迅速升高,随后进入一段时间的弛豫过程,由于Li+自由扩散,电势会逐缓慢下降直到趋于平衡,通过以下公式可以计算出Li+在电极材料内部的扩散系数DLi+。
其中,τ是脉冲电流的持续时间,L对应离子扩散长度,一般指电极活性物质的厚度,ΔEs是相邻两个平衡态的电压差,ΔEτ是恒流充放电引起的电压变化。Li+扩散系数越大,说明电极材料内部的锂离子扩散速度更快,在电极内发生氧化还原过程更快,催化性能更好。GITT还可以实时反映材料充放电过程每一阶段的界面反应电阻信息,对不同充放电阶段的界面反应电阻进行原位监测,用准开路电位和闭路电位之差计算过电位,进而计算瞬态下原位反应电阻:
R = ΔU/(M2 × I)
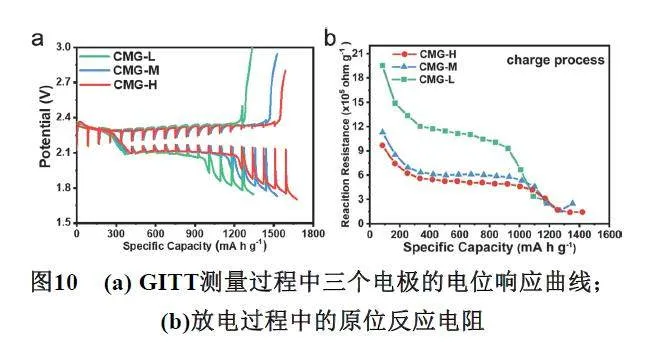
其中,U是准开路电位和闭路电位之差,I是充放电电流密度,M是电极负载质量,如图10所示,越低的瞬态界面反应电阻意味着更快的电荷传质速度,也就意味着电池内部多硫化物转化动力学过程越快81。通常来说,GITT测试虽然可以用来评估原位反应电阻,但是当相应的电化学反应不能维持所需的电流时,就会出现混合电流,在混合电流中,不仅会发生预期的电化学反应,还会发生后续电位较低的反应。因此,混合电流产生时,在GITT数据中会形成下降的线,而不是平台。
4.4 恒电位滴定法
张强团队101将恒电位滴定方法(PotentiostaticIntermittent Titration Technique,PITT)引入锂硫电池体系以评价电池内部电化学性能。以Co-N-C材料和C材料对比为例,具体测试如下:从开路电压开始以每间隔50 mV施加一个恒定的电位,放电至1.8 V,电流对电压脉冲的响应如图11所示。在电化学相变区(2.10–2.05 V,液固转化II)可以明显的识别到电流峰值,表明Li2S1/2 (Li2S和Li2S2的统称)从含有多硫化物的电解液中沉淀出来。对比发现,Co-N-C材料展现出更高的峰值以及更快的响应时间(提前了1650 s),说明Co-N-C材料电池中氧化还原动力学过程更快。此外,在可溶性多硫化物转化区(2.25–2.20 V,Li2Sx-Li2Sx–δ,固液转化I),Co-NC材料的响应电流密度几乎是C材料的3倍,说明了相比于C材料,Co-N-C材料能快速催化多硫化物的转化。
4.5 Li2S沉积实验
单位时间内Li2S的沉积速率以及沉积量也是定量评价材料催化性能的重要因素。具体测试如下:首先将锂硫电池以恒定电流放电至2.06 V,消耗大部分的可溶性多硫化物,施加0.01 V的过电位以诱导Li2S沉淀的驱动力,即在2.05 V的电位下以恒电位模式放电,直到电流低于10−5 A。随后用两个指数函数拟合得到的Li2S8和Li2S6还原的时间-电流曲线,分别拟合相应的参数,计算出不同材料Li2S沉积容量,沉积容量越大说明体系中液-固转化过程越快,材料的催化活性越好(图12) 54。
5 总结
综上所述,锂硫电池中硫还原反应和硫氧化反应动力学缓慢过程引起的穿梭效应是导致锂硫电池尚未商业化应用的重要因素。因此,如何催化多硫化物的快速转化成为关键点。本文不同于以往的材料报道,从电化学角度出发,对多硫化物的存在形式进行了明确的定义,并从吸附-催化、活性中间体两个方面,根据不同的反应机理、路径分析多硫化物转化机制。
吸附-催化根据基底材料与多硫化物之间作用力的强弱可分为物理吸附-催化与化学吸附-催化。传统的多孔碳非极性材料主要是靠物理吸附来锚定多硫化物,通过高导电性来加速电子的转移,促进多硫化物的转化。而化学吸附-催化是指金属化合物,杂原子等掺杂通过形成新的化学键加速多硫化物的裂解。实际上,物理吸附与化学吸附并不是独立存在的,一个优异的电催化剂应当两者兼得,相辅相成,共同促进多硫化物的转化。
锂硫电池体系中由于硫是一系列复杂的多步、多相电化学反应过程,因此活性中间体的形成是不可避免的。不同的活性中间体可能对应着不同的电化学反应路程以及不同的决速步骤。基于此,我们应当大胆假设,仔细求证,找出最佳的反应路线,设计最合理的电催化剂。
最后,本综述总结了目前定量评价锂硫电池催化性能的表征测试手段。以期为锂硫电池电催化剂提供系统全面的设计思路。
References
(1) Ji, X.; Lee, K. T.; Nazar, L. F. Nat. Mater. 2009, 8, 500.doi: 10.1038/nmat2460
(2) Liang, C.; Dudney, N. J.; Howe, J. Y. Chem. Mater. 2009, 21, 4724.doi: 10.1021/cm902050j
(3) Elazari, R.; Salitra, G.; Garsuch, A.; Panchenko, A.; Aurbach, D. Adv.Mater. 2011, 23, 5641. doi: 10.1002/adma.201103274
(4) Ji, L.; Rao, M.; Zheng, H.; Zhang, L.; Li, Y.; Duan, W.; Guo, J.;Cairns, E. J.; Zhang, Y. J. Am. Chem. Soc. 2011, 133, 18522.doi: 10.1021/ja206955k
(5) Shi, Z.; Sun, Z.; Cai, J.; Yang, X.; Wei, C.; Wang, M.; Ding, Y.; Sun,J. Adv. Mater. 2021, 33, e2103050. doi: 10.1002/adma.202103050
(6) Sun, Z.; Zhang, J.; Yin, L.; Hu, G.; Fang, R.; Cheng, H. M.; Li, F.Nat. Commun. 2017, 8, 14627. doi: 10.1038/ncomms14627
(7) Li, D.; Han, F.; Wang, S.; Cheng, F.; Sun, Q.; Li, W. C. ACS Appl.Mater. Interfaces 2013, 5, 2208. doi: 10.1021/am4000535
(8) Zheng, Y.; Zheng, S.; Xue, H.; Pang, H. J. Mater. Chem. A 2019, 7,3469. doi: 10.1039/c8ta11075a
(9) Du, Z.; Chen, X.; Hu, W.; Chuang, C.; Xie, S.; Hu, A.; Yan, W.;Kong, X.; Wu, X.; Ji, H.; et al. J. Am. Chem. Soc. 2019, 141, 3977.doi: 10.1021/jacs.8b12973
(10) Xiao, D.; Li, Q.; Zhang, H.; Ma, Y.; Lu, C.; Chen, C.; Liu, Y.; Yuan,S. J. Mater. Chem. A 2017, 5, 24901. doi: 10.1039/c7ta08483h
(11) Zhu, J.; Cao, J.; Cai, G.; Zhang, J.; Zhang, W.; Xie, S.; Wang, J.; Jin,H.; Xu, J.; Ji, H.; et al. Angew. Chem. Int. Ed. 2023, 62, e202214351.doi:10.1002/anie.202214351
(12) Lu, Y. Q.; Wu, Y. J.; Sheng, T.; Peng, X. X.; Gao, Z. G.; Zhang, S. J.;Deng, L.; Nie, R.; Swiatowska, J.; Li, J. T.; et al. ACS Appl. Mater.Interfaces 2018, 10, 13499. doi: 10.1021/acsami.8b00915
(13) Deng, D. R.; Xue, F.; Jia, Y. J.; Ye, J. C.; Bai, C. D.; Zheng, M. S.;Dong, Q. F. ACS Nano 2017, 11, 6031. doi: 10.1021/acsnano.7b01945
(14) Zhang, H.; Zhao, Z.; Hou, Y. N.; Tang, Y.; Liang, J.; Liu, X.; Zhang,Z.; Wang, X.; Qiu, J. J. Mater. Chem. A 2019, 7, 9230.doi: 10.1039/c9ta00975b
(15) Zhao, M.; Peng, H. J.; Li, B. Q.; Chen, X.; Xie, J.; Liu, X.; Zhang,Q.; Huang, J. Q. Angew. Chem. Int. Ed. 2020, 59, 9011.doi: 10.1002/anie.202003136
(16) Zhang, Y.; Mu, Z.; Yang, C.; Xu, Z.; Zhang, S.; Zhang, X.; Li, Y.; Lai,J.; Sun, Z.; Yang, Y.; et al. Adv. Funct. Mater. 2018, 28, 1707578.doi: 10.1002/adfm.201707578
(17) Zhang, D.; Wang, S.; Hu, R.; Gu, J.; Cui, Y.; Li, B.; Chen, W.; Liu,C.; Shang, J.; Yang, S. Adv. Funct. Mater. 2020, 30, 2002471.doi: 10.1002/adfm.202002471
(18) Wang, Z.; Shen, J.; Liu, J.; Xu, X.; Liu, Z.; Hu, R.; Yang, L.; Feng,Y.; Liu, J.; Shi, Z.; et al. Adv. Mater. 2019, 31, e1902228.doi: 10.1002/adma.201902228
(19) Zhou, J.; Liu, X.; Zhu, L.; Zhou, J.; Guan, Y.; Chen, L.; Niu, S.; Cai,J.; Sun, D.; Zhu, Y.; et al. Joule 2018, 2, 2681.doi: 10.1016/j.joule.2018.08.010
(20) Li, R.; Shen, H.; Pervaiz, E.; Yang, M. Chem. Eng. J. 2021, 404,126462. doi: 10.1016/j.cej.2020.126462
(21) Sul, H.; Bhargav, A.; Manthiram, A. Adv. Energy Mater. 2022, 12,2200680. doi:10.1002/aenm.202200680
(22) Zhao, Q.; Zhu, Q.; Liu, Y.; Xu, B. Adv. Funct. Mater. 2021, 31,2100457. doi: 10.1002/adfm.202100457
(23) Yang, H.; Guo, C.; Chen, J.; Naveed, A.; Yang, J.; Nuli, Y.; Wang, J.Angew. Chem. Int. Ed. 2019, 58, 791. doi: 10.1002/anie.201811291
(24) Mikhaylik, Y. V.; Akridge, J. R. J. Electrochem. Soc. 2004, 151,A1969. doi: 10.1149/1.1806394
(25) Urbonaite, S.; Poux, T.; Novák, P. Adv. Energy Mater. 2015, 5,1500118. doi: 10.1002/aenm.201500118
(26) Gao, Y.; Guo, Q.; Zhang, Q.; Cui, Y.; Zheng, Z. Adv. Energy Mater.2021, 11, 2002580. doi: 10.1002/aenm.202002580
(27) Deng, N.; Liu, Y.; Li, Q.; Yan, J.; Lei, W.; Wang, G.; Wang, L.; Liang,Y.; Kang, W.; Cheng, B. Energy Storage Mater. 2019, 23, 314.doi: 10.1016/j.ensm.2019.04.042
(28) Rosenman, A.; Markevich, E.; Salitra, G.; Aurbach, D.; Garsuch, A.;Chesneau, F. F. Adv. Energy Mater. 2015, 5, 1500212.doi: 10.1002/aenm.201500212
(29) Urbonaite, S.; Novák, P. J. Power Sources 2014, 249, 497.doi: 10.1016/j.jpowsour.2013.10.095
(30) Gao, X.; Sun, Q.; Yang, X.; Liang, J.; Koo, A.; Li, W.; Liang, J.;Wang, J.; Li, R.; Holness, F. B.; et al. Nano Energy 2019, 56, 595.doi: 10.1016/j.nanoen.2018.12.001
(31) Meini, S.; Elazari, R.; Rosenman, A.; Garsuch, A.; Aurbach, D.J. Phys. Chem. Lett. 2014, 5, 915. doi: 10.1021/jz500222f
(32) Rosenman, A.; Elazari, R.; Salitra, G.; Markevich, E.; Aurbach, D.;Garsuch, A. J. Electrochem. Soc. 2015, 162, A470.doi: 10.1149/2.0861503jes
(33) Wang, P.; Xi, B.; Huang, M.; Chen, W.; Feng, J.; Xiong, S. Adv.Energy Mater. 2021, 11. doi: 10.1002/aenm.202002893
(34) Sun, Z.; Vijay, S.; Heenen, H. H.; Eng, A. Y. S.; Tu, W.; Zhao, Y.;Koh, S. W.; Gao, P.; Seh, Z. W.; Chan, K.; et al. Adv. Energy Mater.2020, 10, 1904010. doi: 10.1002/aenm.201904010
(35) Zhang, H.; Tian, D.; Zhao, Z.; Liu, X.; Hou, Y. N.; Tang, Y.; Liang, J.;Zhang, Z.; Wang, X.; Qiu, J. Energy Storage Mater. 2019, 21, 210.doi: 10.1016/j.ensm.2018.12.005
(36) Lim, W. G.; Kim, S.; Jo, C.; Lee, J. Angew. Chem. Int. Ed. 2019, 58,18746. doi: 10.1002/anie.201902413
(37) Liu, S.; Yao, L.; Zhang, Q.; Li, L. L.; Hu, N. T.; Wei, L. M.; Wei, H.Acta Phys. -Chim. Sin. 2017, 33, 2339. [刘帅, 姚路, 章琴, 李路路,胡南滔, 魏良明, 魏浩. 物理化学学报, 2017, 33, 2339.]doi: 10.3866/PKU.WHXB201706021
(38) Wang, J. J. ; Cao, G. Q.; Duan, R. X.; Li, X. Y.; Li, X. F. ActaPhys. -Chim. Sin. 2023, 39, 2212005. [王晶晶, 曹贵强, 段瑞贤,李向阳, 李喜飞. 物理化学学报, 2023, 39, 2212005.]doi: 10.3866/PKU.WHXB202212005
(39) Zhang, M. D.; Chen, B.; Wu, M. B. Acta Phys. -Chim. Sin. 2022, 38,2101001. [张梦迪, 陈蓓, 吴明铂. 物理化学学报, 2022, 38,2101001.] doi: 10.3866/PKU.WHXB202101001
(40) Liu, Y.; Zhang, S.; Qin, X.; Kang, F.; Chen, G.; Li, B. Nano Lett.2019, 19, 4601. doi: 10.1021/acs.nanolett.9b01567
(41) Li, Y.; Zhan, H.; Liu, S.; Huang, K.; Zhou, Y. J. Power Sources 2010,195, 2945. doi: 10.1016/j.jpowsour.2009.11.004
(42) Sun, K.; Zhang, Q.; Bock, D. C.; Tong, X.; Su, D.; Marschilok, A. C.;Takeuchi, K. J.; Takeuchi, E. S.; Gan, H. J. Electrochem. Soc. 2017,164, A1291. doi: 10.1149/2.1631706jes
(43) Wang, W. P.; Zhang, J.; Yin, Y. X.; Duan, H.; Chou, J.; Li, S. Y.;Yan, M.; Xin, S.; Guo, Y. G. Adv. Mater. 2020, 32, e2000302.doi: 10.1002/adma.202000302
(44) Cao, G.; Duan, R.; Li, X. J. Energy Chem 2023, 5, 100096.doi: 10.1016/j.enchem.2022.100096
(45) Martin, R.; Doub, W., Jr.; Roberts, J., Jr.; Sawyer, D. Inorg. Chem.1973, 4, 1921. doi: 10.1002/chin.197339037
(46) Rajput, N. N.; Murugesan, V.; Shin, Y.; Han, K. S.; Lau, K. C.; Chen,J.; Liu, J.; Curtiss, L. A.; Mueller, K. T.; Persson, K. A. Chem. Mater.2017, 29, 3375. doi: 10.1021/acs.chemmater.7b00068
(47) Zhang, B.; Wu, J.; Gu, J.; Li, S.; Yan, T.; Gao, X. P. ACS Energy Lett.2021, 6, 537. doi: 10.1021/acsenergylett.0c02527
(48) Song, Y. W.; Shen, L.; Yao, N.; Li, X. Y.; Bi, C. X.; Li, Z.; Zhou, M.Y.; Li, B. Q.; Huang, J. Q.; Zhang, Q. Chem 2022, 8, 3031.doi: 10.1016/j.chempr.2022.07.004
(49) Luo, Y.; Fang, Z.; Duan, S.; Wu, H.; Liu, H.; Zhao, Y.; Wang, K.;Li, Q.; Fan, S.; Wang, J.; et al. Angew. Chem. Int. Ed. 2023, 62,e202215802. doi:10.1002/anie.202215802
(50) Zheng, S.; Wen, Y.; Zhu, Y.; Han, Z.; Wang, J.; Yang, J.; Wang, C.Adv. Energy Mater. 2014, 4, 1400482. doi: 10.1002/aenm.201400482
(51) Wang, Q.; Zheng, J.; Walter, E.; Pan, H.; Lv, D.; Zuo, P.; Chen, H.;Deng, Z. D.; Liaw, B. Y.; Yu, X. J. Electrochem. Soc. 2015, 162,A474. doi: 10.1149/2.0851503jes
(52) Zhang, G.; Peng, H. J.; Zhao, C. Z.; Chen, X.; Zhao, L. D.; Li, P.;Huang, J. Q.; Zhang, Q. Angew. Chem. Int. Ed. 2018, 57, 16732.doi: 10.1002/anie.201810132
(53) Hu, J.; Long, G.; Liu, S.; Li, G.; Gao, X. Chem. Commun. 2014, 50,14647. doi: 10.1039/C4CC06666A
(54) Yao, W.; Tian, C.; Yang, C.; Xu, J.; Meng, Y.; Manke, I.; Chen, N.;Wu, Z.; Zhan, L.; Wang, Y.; et al. Adv. Mater. 2022, 34, e2106370.doi: 10.1002/adma.202106370
(55) Wang, R.; Yang, J.; Chen, X.; Zhao, Y.; Zhao, W.; Qian, G.; Li, S.;Xiao, Y.; Chen, H.; Ye, Y.; et al. Adv. Energy Mater. 2020, 10,1903550. doi: 10.1002/aenm.201903550
(56) Qiao, Z.; Zhang, Y.; Meng, Z.; Xie, Q.; Lin, L.; Zheng, H.; Sa, B.;Lin, J.; Wang, L.; Peng, D. L. Adv. Funct. Mater. 2021, 31, 2100970.doi: 10.1002/adfm.202100970
(57) Yu, H.; Zhang, B.; Sun, F.; Jiang, G.; Zheng, N.; Xu, C.; Li, Y. Appl.Surf. Sci. 2018, 450, 364. doi: 10.1016/j.apsusc.2018.04.123
(58) Wang, J.; Yang, J.; Wan, C.; Du, K.; Xie, J.; Xu, N. Adv. Funct.Mater. 2003, 13, 487. doi: 10.1002/adfm.200304284
(59) Zhang, Q.; Wang, Y.; Seh, Z. W.; Fu, Z.; Zhang, R.; Cui, Y. NanoLett. 2015, 15, 3780. doi: 10.1021/acs.nanolett.5b00367
(60) Chen, X.; Peng, H. J.; Zhang, R.; Hou, T. Z.; Huang, J. Q.; Li, B.;Zhang, Q. ACS Energy Lett. 2017, 2, 795.doi: 10.1021/acsenergylett.7b00164
(61) Tao, X.; Wang, J.; Liu, C.; Wang, H.; Yao, H.; Zheng, G.; Seh, Z. W.;Cai, Q.; Li, W.; Zu, C. X.; et al. Nat. Commun. 2016, 7, 11203.doi: 10.1038/ncomms11203
(62) Fu, A.; Wang, C.; Pei, F.; Cui, J.; Fang, X.; Zheng, N. Small 2019, 15,1804786. doi: 10.1002/smll.201804786
(63) Peng, X. X.; Lu, Y. Q.; Zhou, L. L.; Sheng, T.; Shen, S. Y.; Liao, H.G.; Huang, L.; Li, J. T.; Sun, S. G. Nano Energy 2017, 32, 503.doi: 10.1016/j.nanoen.2016.12.060
(64) Tao, Y.; Wei, Y.; Liu, Y.; Wang, J.; Qiao, W.; Ling, L.; Long, D. H.Energy Environ. Sci. 2016, 9, 3230. doi: 10.1039/C6EE01662F
(65) Pang, Q.; Nazar, L. F. ACS Nano 2016, 10, 4111.doi: 10.1021/acsnano.5b07347
(66) Liu, J.; Li, W.; Duan, L.; Li, X.; Ji, L.; Geng, Z.; Huang, K.; Lu, L.;Zhou, L.; Liu, Z. R. Nano Lett. 2015, 15, 5137.doi: 10.1021/acs.nanolett.5b01919
(67) Ma, F.; Liang, J.; Wang, T.; Chen, X.; Fan, Y.; Hultman, B.; Xie, H.;Han, J.; Wu, G.; Li, Q. Nano Lett. 2018, 10, 5634.doi: 10.1021/acsnano.0c03325
(68) Wang, C.; Li, K.; Zhang, F.; Wu, Z.; Sun, L.; Wang, L. M. ACS Appl.Mater. Interfaces 2018, 10, 42286. doi: 10.1021/acsami.8b15176
(69) Liu, Y. T.; Han, D. D.; Wang, L.; Li, G. R.; Liu, S.; Gao, X. P. Adv.Energy Mater. 2019, 9, 1803477. doi: 10.1002/aenm.201803477
(70) Zheng, C.; Niu, S.; Lv, W.; Zhou, G.; Li, J.; Fan, S.; Deng, Y.; Pan,Z.; Li, B.; Kang, F.; Yang, Q. H. Nano Energy 2017, 33, 306.doi: 10.1016/j.nanoen.2017.01.040
(71) Liu, G.; Wang, W.; Zeng, P.; Yuan, C.; Wang, L.; Li, H.; Zhang, H.;Sun, X.; Dai, K.; Mao, J.; et al.. Nano Lett. 2022, 22, 6366.doi: 10.1021/acs.nanolett.2c02183
(72) Hou, T. Z.; Xu, W. T.; Chen, X.; Peng, H. J.; Huang, J. Q.; Zhang, Q.Angew. Chem. Int. Ed. 2017, 56, 8178. doi: 10.1002/anie.201704324
(73) Chen, X.; Bai, Y. K.; Zhao, C. Z.; Shen, X.; Zhang, Q. Angew. Chem.Int. Ed. 2020, 59, 11192. doi: 10.1002/anie.201915623
(74) Evers, S.; Yim, T.; Nazar, L. F. J. Phys. Chem. C 2012, 116, 19653.doi: 10.1021/jp304380j
(75) Zhang, M.; Chen, W.; Xue, L.; Jiao, Y.; Lei, T.; Chu, J.; Huang, J.;Gong, C.; Yan, C.; Yan, Y. Adv. Energy Mater. 2020, 10, 1903008.doi: 10.1002/aenm.201903008
(76) Hong, X.; Wang, R.; Liu, Y.; Fu, J.; Liang, J.; Dou, S. J. EnergyChem. 2020, 42, 144. doi: 10.1016/j.jechem.2019.07.001
(77) Wang, X.; Gao, T.; Han, F.; Ma, Z.; Zhang, Z.; Li, J.; Wang, C. NanoEnergy 2016, 30, 700. doi: 10.1016/j.nanoen.2016.10.049
(78) Wang, Y.; Zhu, L.; Wang, J.; Zhang, Z.; Yu, J.; Yang, Z. Chem. Eng. J.2022, 433, 133792. doi: 10.1016/j.cej.2021.133792
(79) Zhu, Y.; Wang, S.; Miao, Z.; Liu, Y.; Chou, S. L. Small 2018, 14,1801987. doi: 10.1002/smll.201801987
(80) Barchasz, C.; Molton, F.; Duboc, C.; Lepretre, J. C.; Patoux, S.;Alloin, F. Anal. Chem. 2012, 84, 3973. doi: 10.1021/ac2032244
(81) Xu, R.; Tang, H.; Zhou, Y.; Wang, F.; Wang, H.; Shao, M.; Li, C.;Wei, Z. D. Chem. Sci. 2022, 13, 6224. doi: 10.1039/d2sc01353c
(82) Cuisinier, M.; Hart, C.; Balasubramanian, M.; Garsuch, A.; Nazar, L.F. Adv. Energy Mater. 2015, 5, 1401801.doi: 10.1002/aenm.201401801
(83) Tong, C.; Chen, H.; Jiang, S.; Li, L.; Shao, M.; Li, C.; Wei, Z. ACSAppl. Mater. Interfaces 2023, 15, 1175. doi: 10.1021/acsami.2c18594
(84) Wujcik, K. H.; Pascal, T. A.; Pemmaraju, C.; Devaux, D.; Stolte, W.C.; Balsara, N. P.; Prendergast, D. Adv. Energy Mater. 2015, 5,1500285. doi: 10.1002/aenm.201500285
(85) Wang, C.; Ma, Y.; Du, X.; Zhang, H.; Xu, G.; Cui, G. Battery Energy2022, 1, 20220010. doi: 10.1002/bte2.20220010
(86) Liang, X.; Kwok, C. Y.; Lodi-Marzano, F.; Pang, Q.; Cuisinier, M.;Huang, H.; Hart, C. J.; Houtarde, D.; Kaup, K.; Nazar, L. F.; et al.Adv. Energy Mater. 2016, 6, 1501636. doi: 10.1002/aenm.201501636
(87) Wang, S.; Liao, J.; Yang, X.; Liang, J.; Sun, Q.; Liang, J.; Zhao, F.;Koo, A.; Kong, F.; Sun, X. L.; et al. Nano Energy 2019, 57, 230.doi: 10.1016/j.nanoen.2018.12.020
(88) Hua, W.; Li, H.; Pei, C.; Xia, J.; Sun, Y.; Zhang, C.; Lv, W.; Tao, Y.;Jiao, Y.; Zhang, B.; et al. Adv. Mater. 2021, 33, e2101006.doi: 10.1002/adma.202101006
(89) Wang, J.; Jia, L.; Zhong, J.; Xiao, Q.; Wang, C.; Zang, K.; Liu, H.;Zheng, H.; Luo, J.; Yang, J.; et al. Energy Storage Mater. 2019, 18,246. doi: 10.1016/j.ensm.2018.09.006
(90) Feng, J.; Yi, H.; Lei, Z.; Wang, J.; Zeng, H.; Deng, Y.; Wang, C.J. Energy Chem. 2021, 56, 171. doi: 10.1016/j.jechem.2020.07.060
(91) Li, Z.; Zhang, S.; Zhang, C.; Ueno, K.; Yasuda, T.; Tatara, R.;Dokko, K.; Watanabe, M. Nanoscale 2015, 7, 14385.doi: 10.1039/C5NR03201F
(92) Chen, S.; Dai, F.; Gordin, M. L.; Yu, Z.; Gao, Y.; Song, J.; Wang, D.Angew. Chem. Int. Ed. 2016, 55, 4231. doi: 10.1002/anie.201511830
(93) Zhao, M.; Li, B. Q.; Chen, X.; Xie, J.; Yuan, H.; Huang, J. Q. Chem2020, 6, 3297. doi: 10.1016/j.chempr.2020.09.015
(94) Li, G.; Wang, X.; Seo, M. H.; Li, M.; Ma, L.; Yuan, Y.; Wu, T.; Yu,A.; Wang, S.; Lu, J.; et al. Nat. Commun. 2018, 9, 705.doi: 10.1038/s41467-018-03116-z
(95) Zhao, C. X.; Li, X. Y.; Zhao, M.; Chen, Z. X.; Song, Y. W.; Chen,W. J.; Liu, J. N.; Wang, B.; Zhang, X. Q.; Chen, C. M.; et al. J. Am.Chem. Soc. 2021, 143, 19865. doi: 10.1021/jacs.1c09107
(96) Guo, C.; Liu, M.; Gao, G. K.; Tian, X.; Zhou, J.; Dong, L. Z.; Li,Q.; Chen, Y.; Li, S. L.; Lan, Y. Q. Angew. Chem. Int. Ed. 2022, 134,e202113315. doi: 10.1002/ange.202113315
(97) Kaiser, M. R.; Chou, S.; Liu, H. K.; Dou, S. X.; Wang, C.; Wang, J.Adv. Mater. 2017, 29, 1700449. doi: 10.1002/adma.201700449
(98) Lin, Y.; Huang, S.; Zhong, L.; Wang, S.; Han, D.; Ren, S.; Xiao, M.;Meng, Y. Energy Storage Mater. 2021, 34, 128.doi: 10.1016/j.ensm.2020.09.009
(99) Gao, H.; Ning, S.; Lin, J.; Kang, X. Energy Storage Mater. 2021, 40,312. doi: 10.1016/j.ensm.2021.05.027
(100) Dong, Q.; Wang, T.; Gan, R.; Fu, N.; Li, C.; Wei, Z. ACS Appl.Mater. Interfaces 2020, 12, 20596. doi: 10.1021/acsami.0c04554
(101) Li, B. Q.; Kong, L.; Zhao, C. X.; Jin, Q.; Chen, X.; Peng, H. J.; Qin,J. L.; Chen, J. X.; Yuan, H.; Zhang, Q.; Huang, J. Q. InfoMat 2019,1, 533. doi: 10.1002/inf2.12056
国家自然科学基金(22075033, U21A20312, 91834301)资助项目
猜你喜欢
物理化学学报(2024年9期)2024-09-27 00:00:00
石油化工技术与经济(2023年4期)2023-09-16 04:50:04
无机化学学报(2018年4期)2018-04-10 09:26:49
中国有色金属学报(2018年2期)2018-03-26 07:58:37
分析化学(2018年12期)2018-01-22 12:31:46
环境保护与循环经济(2017年1期)2017-09-26 11:44:27
中南大学学报(自然科学版)(2016年2期)2017-01-19 07:37:25
中国资源综合利用(2016年7期)2016-02-03 03:00:13
电源技术(2015年7期)2015-08-22 08:48:28
物理化学学报(2015年5期)2015-02-28 17:34:59
