
一种有效的黑曲霉启动子捕获探针的构建及其应用
2024-05-16 03:34彭亚彬徐泽宇韩天琦王丽娟薛鲜丽王德培
食品研究与开发 2024年9期
彭亚彬,徐泽宇,韩天琦,王丽娟,薛鲜丽,2,王德培,3*
(1.天津科技大学生物工程学院,天津 300450;2.天津市微生物代谢与发酵过程控制技术工程中心,天津 300450;3.天津工业发酵微生物教育部重点实验室,天津 300457)
黑曲霉作为一种食品安全性的丝状真菌,已成为国际公认的重要工业生产菌株[1]。由于其具有极强的活性蛋白分泌以及蛋白质翻译后的加工能力,成为发酵生产主要菌种之一,广泛应用于各种酶制剂的生产、食品加工、生物肥料、废物处理、药物生产等领域[2-3]。尽管黑曲霉是大多数发酵产品的理想来源,并且作为宿主已经成功表达多种蛋白,但是仍然存在发酵产量达不到工业生产要求水平的问题,如何实现目的基因在黑曲霉中高效表达,是提高发酵产量的关键。目前基因表达调控方式主要包括DNA 水平调控[4]、转录水平调控[5]、翻译水平调控[6]、翻译后修饰调控及蛋白质转运调控[7-8]等,其中转录调控是黑曲霉及其开发利用研究的热点,通过筛选或改造黑曲霉的启动子可显著提高目的基因的转录水平[9-10]。
启动子是一段位于结构基因上游5′端DNA 序列,作为顺式作用元件发挥着重要作用,参与基因转录的起始,通过与转录因子结合来调控基因转录活性[11]。黑曲霉具有复杂的菌丝形态与异核体等问题,缺乏快速准确的启动子评估方法,导致目前用于构建黑曲霉细胞工厂构建的强启动子有限,从而限制了黑曲霉的开发利用。因此筛选更多高效表达的启动子元件对于实现黑曲霉合成各类发酵产品是非常重要的。目前真菌启动子的研究方法主要有利用启动子探针质粒载体筛选启动子、利用聚合酶链式反应(polymerase chain reaction,PCR)技术克隆启动子、基因组文库筛选法和利用载体或接头的染色体步移技术克隆启动子[12]等,而荧光蛋白基因广泛地被用作报告基因来表征启动子的启动效果[8,12-14]。获得启动子序列后,可根据已报道的黑曲霉启动子关键元件对其进行序列分析[15-16],除此之外通过生物信息学的方法对启动子进行分析推测也是一种重要的方法[17-18]。
本研究构建一种具有红色荧光蛋白(Rfp)和潮霉素抗性(HygR)基因表达框的启动子捕获探针载体,Rfp基因无终止子,HygR基因带有构巢曲霉PtrpC启动子可自主表达。通过农杆菌转化法将表达框随机整合到黑曲霉基因组中,以潮霉素抗性筛选出突变株[19],根据红色荧光效果获得启动活性强的启动子,通过交错热不对称链式聚合酶反应获得Rfp上游未知启动子序列[20],并以5′端缺失法对获得的启动子进行鉴定[21-22],以期为丝状真菌强启动子筛选提供一种可参考的方法。
1 材料与方法
1.1 材料
大肠杆菌菌株DH5α、农杆菌菌株EHA105、黑曲霉菌株CGMCC 10142、pN1 质粒[有红色荧光蛋白基因(Rfp)、潮霉素抗性基因(HygR)和卡那霉素抗性基因(KanaR)]、p40 质粒(在大肠杆菌和根癌农杆菌中稳定复制带有KanaR):天津科技大学生化过程与控制实验室保存;潮霉素B、抗生素卡那霉素、氨苄青霉素、噻孢霉素钠:北京索来宝科技有限公司;Primer Star Max酶、限制性内切酶、DNA 连接酶、Genome Walking Kit试剂盒、10 000 bp DNA marker、5 000 bp DNA marker:北京宝日医生物技术有限公司;质粒小提试剂盒、琼脂糖凝胶回收试剂盒:天根生化科技有限公司;2×Rapid Taq 酶:南京诺维赞生物科技有限公司;所有的引物均由金唯智生物科技有限公司合成,所用引物序列见表1。

表1 试验用到的引物Table 1 Primers used in this study
1.2 培养基
1.3 主要仪器
ME104 电子天平:上海梅特勒-托利多仪器有限公司;SW-CJ-ID 超净工作台:苏州净化设备有限公司;TCL 台式高速冷冻离心机:中科院生物物理所技术公司;Champ Gel 6000 全自动凝胶成像仪:北京赛智创业科技有限公司;DYY-6C 电泳仪:北京六一仪器厂;Mastercycler X50PCR 仪:上海恒久医疗器械有限公司;SPX-250B-Z 生化培养箱:上海博迅实业有限公司医疗设备厂;BX50 正置荧光显微镜:上海奥林巴斯贸易有限公司。
1.4 方法
1.4.1 PCR 反应体系和反应条件
PCR 反应体系和反应条件见表2。
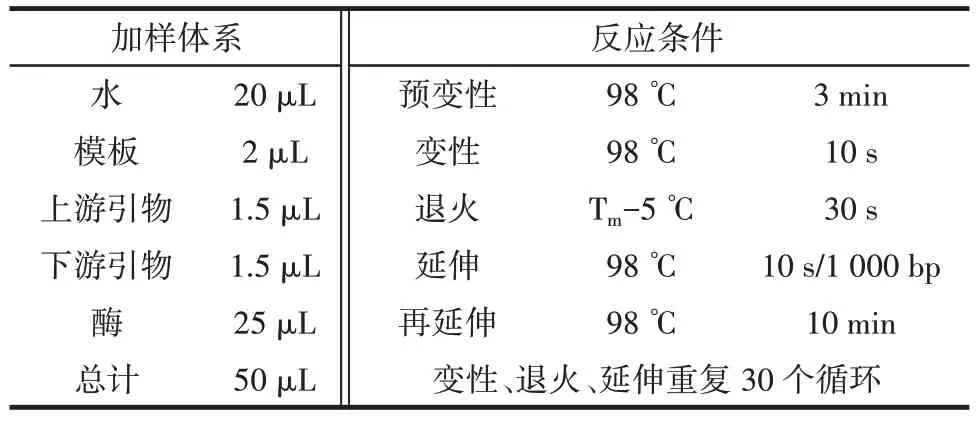
表2 PCR 反应体系和反应条件Table 2 PCR reaction system and conditions
1.4.2 启动子捕获探针质粒载体的构建
在此次研究中,我们对70例股骨粗隆骨折患者进行了分析,均是老年患者,为患者提供了针对性的护理,提供镇痛和深静脉血栓的护理,避免患者在接受治疗的过程中出现并发症,根据研究结果显示,全部患者接受护理之后,共有65例顺利的度过了围手术期,有5例患者在治疗阶段有并发症产生,经过护理人员针对性的积极的护理,患者均痊愈,全部患者顺利康复出院。
启动子捕获探针质粒载体pN3 的构建方法见图1。
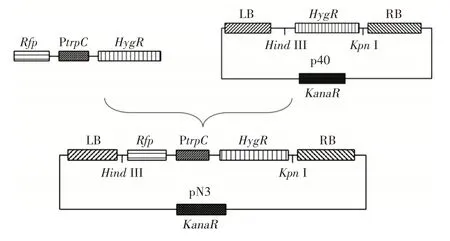
图1 pN3 质粒构建Fig.1 Construction of pN3 plasmid
由图1 可知,首先以pN1 质粒为模板,设计带有酶切位点的接头引物Rfp-C-F 和Hyg-C-R 扩增Rfp-PtrpC-HygR片段,进行琼脂糖凝胶电泳并根据试剂盒说明书切胶回收;对切胶回收产物即回收片段和p40质粒分别用限制性内切酶HindШ、KpnI 进行双酶切,再用连接酶进行连接,将连接产物转化大肠杆菌DH5α,筛选阳性克隆酶切验证并进行测序,将启动子捕获探针质粒载体命名为pN3。
1.4.3 根癌农杆菌介导的黑曲霉转化及突变株的筛选
将含有目的片段的质粒使用电转化法转入农杆菌EHA105 感受态细胞中,经卡那霉素抗性筛选单菌落并通过PCR 验证,将正确转化子与黑曲霉CGMCC 10142 共同培养,将2×107个/mL 的新鲜黑曲霉孢子与100 μL 的OD600=0.8 的验证正确的农杆菌转化子混合,涂布于0.45 μm 的Hybond N+滤膜上,置于IM 培养基表面,25 ℃培养箱共培养48 h。将共培养完成后的菌体洗脱至含有150 μg/mL 潮霉素B 的CM 平板进行初步筛选,35 ℃培养箱培养2~3 d,长出的单菌落可能为获得Rfp-PtrpC-HygR片段插入的突变株,将这些单菌落挑取转接至潮霉素浓度更高的CM 平板复筛。
1.4.4 黑曲霉突变株的荧光测定
将黑曲霉菌株于CM 培养基上37 ℃培养24 h,黑曲霉突变株M-pglaA 于糖诱导条件下培养,挑取CM培养基上适量的突变株幼嫩菌丝至载玻片,放置在正置荧光显微镜的载物台上,高倍镜观察菌丝形态。在黑暗环境中找到清晰的菌丝后选择绿色激发光观察[14]。
1.4.5 黑曲霉突变株Rfp 基因上游序列的获取
提取具有红色荧光的黑曲霉突变株的基因组,PCR 验证插入片段Rfp-PtrpC-HygR正确后,再以该突变株基因组为模板,通过交错热不对称链式聚合酶反应扩增出插入片段上游未知启动子序列,并进行测序;根据测序结果设计引物P1-F 和P1-R 验证启动子和Rfp-PtrpC-HygR片段的共线性;将测序结果与Rfp序列进行比对,获得未知启动子序列;将获得的序列于NCBI 数据库比对,确定该启动子所启动表达的基因,选取该基因上游启动子区作为进一步研究对象。
1.4.6 启动子5′端缺失质粒构建
Pgh启动子5′端缺失质粒和pglaA 质粒构建思路如图2 所示。
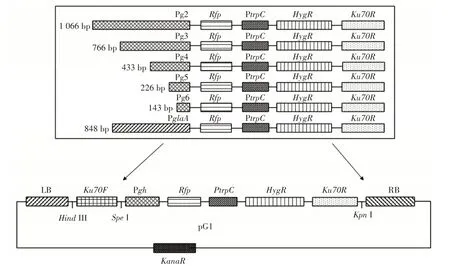
图2 Pgh 启动子5′端缺失质粒和pglaA 质粒构建Fig.2 Construction of Pgh 5′deletion and pglaA plasmids
构建Pgh启动子5′端缺失质粒思路如下:首先构建pG1 质粒即包括完整的Pgh启动子,以Ku70基因序列为同源臂,中间插入Pgh启动子、Rfp基因和HygR基因。pG1 质粒构建成功后,以其为母本继续构建pG2、pG3、pG4、pG5、pG6 质粒、依次对Pgh启动子进行5′端缺失。pG1 质粒具体构建方法如下:以黑曲霉菌株CGMCC 10142 基因组为模板,以引物Ku70F-F和Ku70F-R 扩增Ku70F;以引物Ku70R-F 和Ku70R-R扩增Ku70R;以引物K-PgH-F 和PgH-r-R 扩增Pgh启动子;以pN3 质粒为模板,以引物P-Rfp-F 和HYG-KR 扩增片段Rfp-PtrpC-HygR。将扩增得到的Ku70F和Pgh启动子通过PCR 融合获得片段ku70F-Pgh,再将获得的Rfp-PtrpC-HygR和Ku70R通过PCR 融合获得片段Rfp-PtrpC-HygR-Ku70R,最后将两片段通过融合PCR 获得最终片段。将该目的片段和p40 质粒通过BamHI 和HindШ 进行双酶切连接,转化至大肠杆菌DH5α,PCR 和双酶切验证正确后用于pG2、pG3、pG4、pG5、pG6 和pglaA 质粒的构建。分别以pG1 质粒为模板,扩增Pg2/Pg3/Pg4/Pg5/Pg6-Rfp-PtrpC-HygRKu70R,SpeI 和KpnI 双酶切pG1 质粒作为载体、Pg2/Pg3/Pg4/Pg5/Pg6-Rfp-PtrpC-HygR-Ku70R并使用T4连接酶连接载体和片段,化转至大肠杆菌DH5α,PCR和双酶切验证正确后用于下一步试验。以出发菌株基因组为模板扩增糖化酶启动子(PglaA:848 bp),与从pG1 质粒扩增的Rfp-PtrpC-HygR-Ku70R融合获得片段PglaA-Rfp-PtrpC-HygR-Ku70R,以相同的方式构建pglaA 质粒。
2 结果与分析
2.1 启动子捕获探针质粒载体pN3 的构建
pN3 质粒探针和其黑曲霉突变株筛选及PCR 验证见图3。
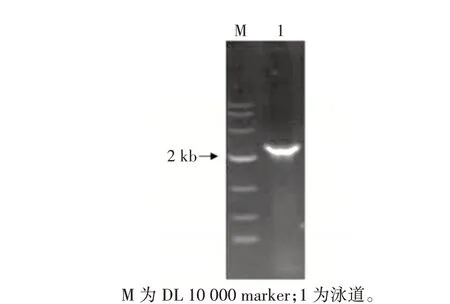
图3 pN3 质粒PCR 验证Fig.3 PCR validation of pN3 plasmid
由图3 可知,按照1.4.2 所述方法构建pN3 质粒,并进行PCR 验证,结果与预期一致,探针质粒pN3 构建完成。
2.2 pN3 质粒黑曲霉突变株的筛选与验证
pN3 质粒黑曲霉突变株的筛选与验证结果见图4。
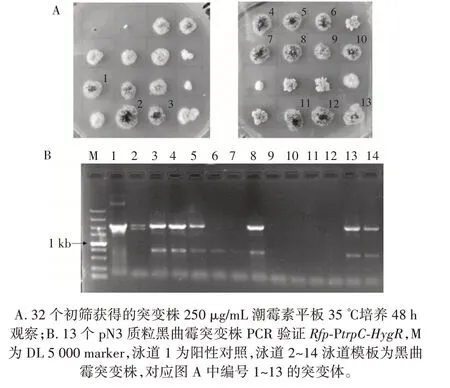
图4 pN3 质粒探针黑曲霉突变株筛选及PCR 验证Fig.4 Screening and PCR validation of Aspergillus niger mutants with pN3 plasmid probe
通过潮霉素抗性对pN3 质粒黑曲霉突变株进行筛选并对筛选到的黑曲霉突变体进行PCR 验证,具体方法如下:提取大肠杆菌DH5α 中的pN3 质粒,电转化至农杆菌EHA105,由其介导整合到黑曲霉菌株CGMCC 10142 基因组中,以150 μg/mL 潮霉素筛选突变株,初步筛选出具有潮霉素抗性的327 个突变株,从这些突变株随机挑选出32 个于250 μg/mL 潮霉素B的CM 培养基平板上35 ℃培养72 h,选取潮霉素抗性强的13 个突变株的基因组进行PCR 验证Rfp-HygR片段,得到7 株PCR 验证正确的突变株,巢氏PCR 扩增HygR片段进一步验证这7 株突变株的正确性,均为正确随机插入突变株。
2.3 pN3 质粒黑曲霉突变体的荧光测定
pN3 质粒黑曲霉突变体的荧光测定结果见图5。
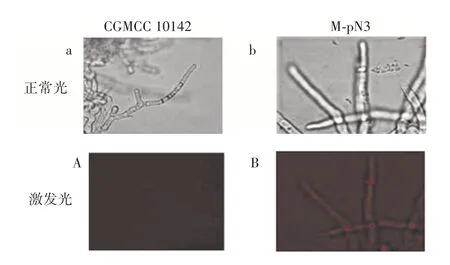
图5 黑曲霉突变株M-pN3 菌丝荧光观察Fig.5 Fluorescence detection of Aspergillus niger mutant M-pN3
对PCR 验证正确的7 株突变株进行正置荧光显微镜观察,发现12 号突变株菌丝具有相对较强的红色荧光特征,其余6 株突变株红色荧光强度都比12 号突变株弱或者观测不到荧光,原因是HygR基因具有启动子,能够自主表达,存在HygR基因随机整合到黑曲霉基因组的可能,出现潮霉素抗性但无红色荧光表型,因此以红色荧光强度作为筛选启动子的报告基因,将12 号突变株其命名为M-pN3。
2.4 M-pN3 菌株报告基因上游启动子的获得与分析
Tail-PCR 扩增M-pN3 突变株Rfp-PtrpC-HygR片段上游未知序列结果见图6。
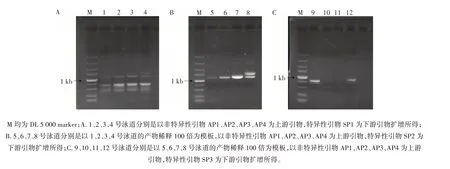
图6 Tail-PCR 扩增M-pN3 突变株Rfp-PtrpC-HygR 片段上游未知序列Fig.6 Amplification of the upstream unknown sequence of Rfp-PtrpC-HygR of M-pN3 by Tail-PCR
以下游特异性引物SP1、SP2、SP3,上游非特异性引物AP1、AP2、AP3、AP4(试剂盒提供,序列未知)通过Tail-PCR 获得M-pN3 基因组插入片段Rfp-PtrpCHygR上游序列,回收第三轮扩增的明亮的条带并进行测序并使用NCBI 数据库比对,发现该段序列与A.nigerCBS513.88 数据库公布的A.nigerAn08g05220 序列(基因ID:AM270168.1)同源性高达100%。根据比对结果追溯其下游序列,通过与A.nigerCBS513.88 数据库比对,得知该序列下游为An08g05230 基因序列,编码糖苷水解酶基因。因此将得到的糖苷水解酶编码基因上游的序列截取1 500 bp 的长度命名为Pgh启动子。
Pgh启动子序列分析见图7。

图7 Pgh 启动子序列分析Fig.7 Sequence analysis of Pgh promoters
将Pgh启动子利用启动子预测软件Promoter 2.0进行预测,并根据已报道的启动子元件序列对Pgh启动子进行分析,发现该启动子有很大的可能性具有启动活性。对Pgh启动子进行分析:Pgh启动子中含有启动子通用元件TATA 框,多个CAAT-box、CT-box、GCbox 和多个诱导型元件,且具有黑曲霉启动子通用序列CTTCTC,具体元件类型和位置见表3。
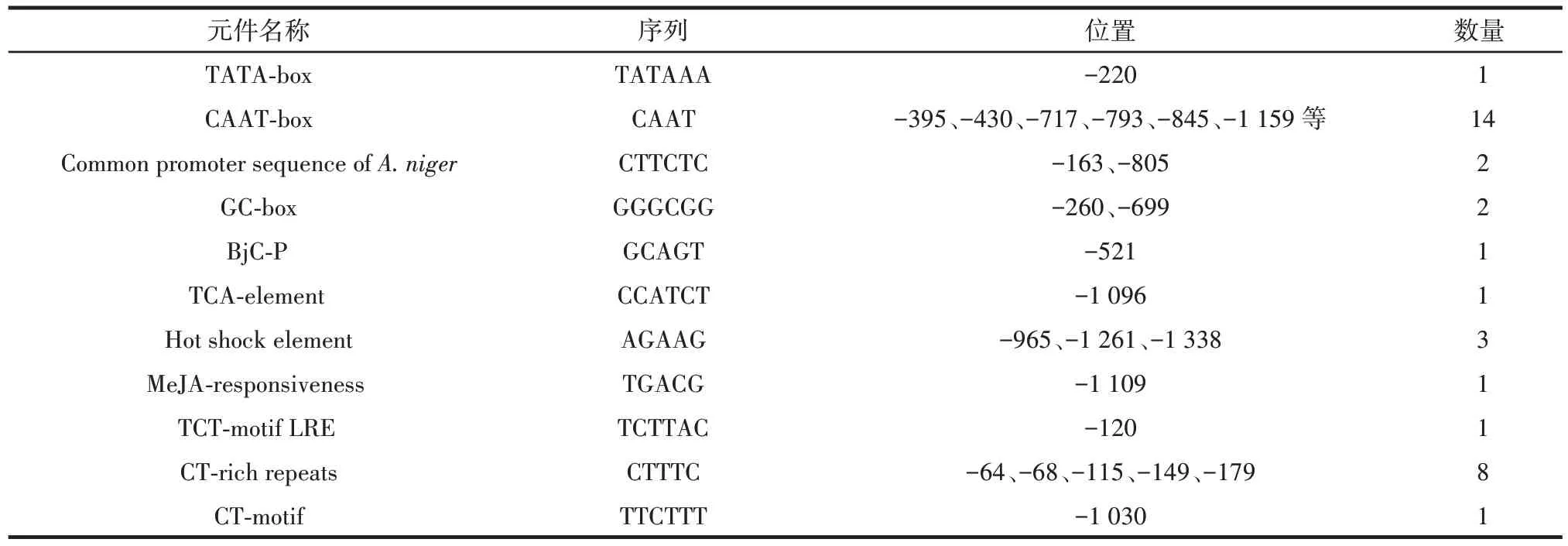
表3 Pgh 启动子元件分析Table 3 Analysis of Pgh promoter components
2.5 Pgh 启动子5′端缺失质粒及pglaA 质粒的构建
首先构建pG1 质粒即包括完整的Pgh启动子,以Ku70基因序列为同源臂,中间插入Pgh启动子、Rfp基因和HygR基因。pG1 质粒构建成功后,以其为模板继续构建pG2、pG3、pG4、pG5 和pG6 质粒,依次对Pgh启动子进行5′端缺失并将pG1 质粒的Pgh启动子替换为PglaA构建pglaA 质粒。
2.6 pG 系列质粒黑曲霉菌株突变株的筛选
pG1~6 质粒黑曲霉突变株的PCR 验证结果见图8。
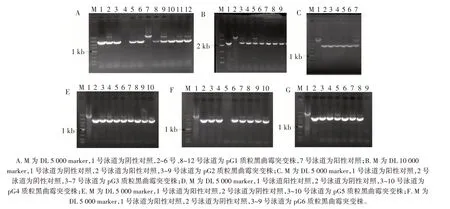
图8 pG1~6 质粒黑曲霉突变株的PCR 验证Fig.8 PCR validation of Aspergillus niger mutants with pG1-6 plasmids
由图8 可知,对获得的质粒pG1~6 的黑曲霉突变体提取基因组进行PCR 验证,验证引物均为Ku70F-F和Ku70R-R,正确的突变株命名为M-pG1~6。
以Ku70为表达框Rfp-PtrpC-HygR的左右臂,构建pG1~6 质粒探针目的是将其表达框同源重组在同一位点上,使试验结果更准确,但是由于黑曲霉获得同源重组突变株异常困难,pG1~6 质粒表达框均未成功整合到Ku70位点。
2.7 pG 系列质粒黑曲霉菌株突变株荧光观察及其潮霉素抗性验证
黑曲霉突变体菌丝荧光观察及其潮霉素抗性验证结果见图9。
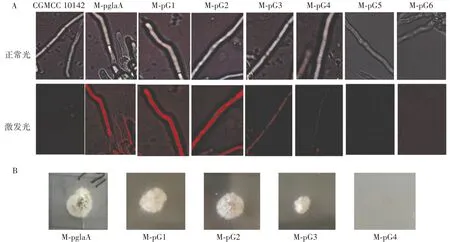
图9 黑曲霉突变体菌丝荧光观察及其潮霉素抗性验证Fig.9 Mycelial fluorescence detection and hygromycin resistance verification of Aspergillus niger mutants
将突变株接种于CM 固体培养基上,35 ℃培养24 h,在正置荧光显微镜下观察菌丝红色荧光,结果如图9A 所示,M-pG1 和M-pG2 突变株菌丝有较强的红色荧光,与对照突变株M-pglaA 的红色荧光强度无显著差异,M-pG3 突变株菌丝红色荧光较弱,M-pG4 突变株菌丝红色荧光微弱,M-pG5 和M-pG6 突变株菌丝未检测到红色荧光,表明pG5 和pG6 序列不具有启动活性或者启动活性极低,将M-pglaA、M-pG1~M-pG6 各黑曲霉突变株成熟孢子点种于250 μg/mL 潮霉素B 的CM 固体培养基上35 ℃培养48 h,结果如图9B 所示。各突变株潮霉素抗性生长情况与红色荧光表达存在极好相关性,其中M-pglaA、M-pG1、M-pG2 红色荧光较强,其抗潮霉素生长情况较好,48 h 菌落直径一致且长出孢子,相对于上述3 株菌M-pG3 红色荧光弱,其抗潮霉素生长情况也表现出菌落小且不产孢子,而MpG4 红色荧光微弱,表现出在250 μg/mL 浓度抗潮霉素条件下无法生长,M-pG5 和M-pG6 突变株完全不萌发故未放置图片。这些结果表明:通过5′端缺失所得到的不同启动子区域序列,其启动表达红色荧光蛋白活性与其潮霉素抗性具有明确正相关性,由此推断潮霉素抗性基因不仅可以作为筛选标记,也可以作为副报告基因来评价黑曲霉启动子的启动效果。
3 讨论
关于利用农杆菌介导的T-DNA 随机插入法捕获启动子,目前仅在水稻和小麦等植物和很少的真菌中有报道[24-25]。本研究构建了Rfp报告基因和HygR筛选标记的质粒探针pN3,通过农杆菌转化法将表达框随机整合到黑曲霉基因组中,通过潮霉素抗性对突变株筛选,再通过PCR 对突变株基因型验证,对验证正确的突变株进行红色荧光观察,获得了一株能在300 μg/mL浓度潮霉素的CM 平板上生长产孢且菌丝红色荧光强的M-pN3 突变株。根据本课题组长期使用潮霉素作为黑曲霉突变体筛选标记的经验,在黑曲霉中,上游基因的启动子会对与其串联的下游基因的表达起到一定的促进作用,启动子的启动活性越强,对下游基因的表达促进作用越大。根据这个发现,设计了启动子捕获探针pN3,通过250 μg/mL 较高浓度潮霉素对初筛获得的突变株复筛,以捕获具有较强启动活性的启动子。本研究中,通过250 μg/mL 潮霉素浓度复筛获得13 株突变体,经PCR 验证有7 株突变株成功整合表达框到基因组中,整合效率为53.8%。
Tail-PCR 法是获得基因组中已知基因序列侧翼序列最常用的方法,陈璨[26]通过农杆菌介导的转化,利用Tail-PCR 技术扩增出多个红曲霉突变体T-DNA 侧翼序列,并通过测序和比对获得这些序列的详细信息。本研究通过Tail-PCR 获得M-pN3 突变株Rfp-PtrpCHygR表达框上游未知启动子序列,将测序结果在NCBI 数据库进行序列比对,确定该序列位于黑曲霉CBS 513.88 糖苷水解酶上游的启动子区,获取该基因起始密码子上游1 500 bp 序列命名为Pgh启动子。为进一步研究Pgh启动子,我们采用了简单而高效的5′端缺失法构建探针质粒pg1~6 对其进一步分析,Xiao等[27]曾利用5′端缺失法分析PsrbB启动子,确定-1 024 ~-588 bp 为其具有缺氧诱导活性的核心区域。根据5′端缺失启动子突变体的红色荧光观察结果,启动子Pgh的-1 066~-766 bp 区域能极大增强启动子启动活性,结合启动子元件分析可以发现这段序列中有较多CAAT-box 和一个CT-motif,这两种元件是丝状真菌启动子重要元件[28];而在-766~-433 bp 和-433~226 bp 间各有一个CAAT-box 和GC-box,而在对照黑曲霉启动子PglaA中同样具有GC-box、多个CAAT-box 和CTmotif,这些结果证明黑曲霉启动子PPgh是一个较强的启动子,CAAT-box、CT motif 和GC-box 是黑曲霉强启动子中必要的转录启动元件。
4 结论
本研究通过启动子捕获探针pN3 筛选出一株具有正确T-DNA 随机插入、菌丝具有较强荧光特征且具有较高潮霉素抗性的突变菌株,通过交错热不对称链式聚合酶反应获得T-DNA 侧翼启动子序列Pgh,并通过5′端缺失法确定了-1 066~-766 bp 区域为其启动核心区域。pN3 启动子探针捕获质粒为黑曲霉及其他丝状真菌天然强启动子捕获提供一个简单的工具,也可用于其他丝状真菌启动子的筛选鉴定,同时为进一步构建黑曲霉杂合启动子奠定基础。
猜你喜欢
种子(2023年4期)2023-06-19
江苏农业科学(2021年1期)2021-03-15
甘肃农业科技(2019年8期)2019-09-10
现代检验医学杂志(2016年3期)2016-11-15
三峡大学学报(自然科学版)(2016年6期)2016-04-16
中国酿造(2016年12期)2016-03-01
中国酿造(2016年12期)2016-03-01
大连工业大学学报(2015年4期)2015-12-11
物理实验(2015年9期)2015-02-28
储能科学与技术(2014年6期)2014-02-27
