
Novel insights into immune-related genes associated with type 2 diabetes mellitus-related cognitive impairment
2024-04-22 00:59:22JingGaoYingZouXiaoYuLvLiChenXinGuoHou
World Journal of Diabetes 2024年4期
Jing Gao,Ying Zou,Xiao-Yu Lv,Li Chen,Xin-Guo Hou
Abstract BACKGROUND The cognitive impairment in type 2 diabetes mellitus (T2DM) is a multifaceted and advancing state that requires further exploration to fully comprehend.Neuroinflammation is considered to be one of the main mechanisms and the immune system has played a vital role in the progression of the disease.AIM To identify and validate the immune-related genes in the hippocampus associated with T2DM-related cognitive impairment.METHODS To identify differentially expressed genes (DEGs) between T2DM and controls,we used data from the Gene Expression Omnibus database GSE125387.To identify T2DM module genes,we used Weighted Gene Co-Expression Network Analysis.All the genes were subject to Gene Set Enrichment Analysis.Protein-protein interaction network construction and machine learning were utilized to identify three hub genes.Immune cell infiltration analysis was performed.The three hub genes were validated in GSE152539 via receiver operating characteristic curve analysis.Validation experiments including reverse transcription quantitative realtime PCR,Western blotting and immunohistochemistry were conducted both in vivo and in vitro.To identify potential drugs associated with hub genes,we used the Comparative Toxicogenomics Database (CTD).RESULTS A total of 576 DEGs were identified using GSE125387.By taking the intersection of DEGs,T2DM module genes,and immune-related genes,a total of 59 genes associated with the immune system were identified.Afterward,machine learning was utilized to identify three hub genes (H2-T24,Rac3,and Tfrc).The hub genes were associated with a variety of immune cells.The three hub genes were validated in GSE152539.Validation experiments were conducted at the mRNA and protein levels both in vivo and in vitro,consistent with the bioinformatics analysis.Additionally,11 potential drugs associated with RAC3 and TFRC were identified based on the CTD.CONCLUSION Immune-related genes that differ in expression in the hippocampus are closely linked to microglia.We validated the expression of three hub genes both in vivo and in vitro,consistent with our bioinformatics results.We discovered 11 compounds associated with RAC3 and TFRC.These findings suggest that they are co-regulatory molecules of immunometabolism in diabetic cognitive impairment.
Key Words: Bioinformatics analysis;Type 2 diabetes mellitus;Cognitive impairment;Hippocampus;Immune;Microglia
INTRODUCTION
The prevalence of type 2 diabetes mellitus (T2DM) is rapidly increasing due to lifestyle changes and the aging population.Over the past few years,an increasing number of researches have emphasized the connection between T2DM and cognitive impairment[1].The severity of cognitive impairment in T2DM may range from mild cognitive decline to more severe forms,including dementia and Alzheimer's disease (AD)[2].Cognitive impairment in T2DM is a progressively advancing condition.Once upon diagnosis,there is no effective treatment currently available.The pathophysiology of the condition is multifactorial and there are still areas yet to be fully investigated.
Immune system abnormalities play a pivotal role in the pathogenesis of T2DM.Dysregulation of inflammation and immune responses is intricately linked to insulin resistance and beta cell dysfunction[3].Furthermore,alterations in the expression of immune-related genes may also serve as crucial determinants in the development of diabetes-associated cognitive impairment.Therefore,a complete understanding of the involvement of these immune genes in the cognitive impairment of individuals with T2DM is imperative.
Bioinformatics is used to screen differences at multiple levels from microarray or high-throughput sequencing data,between patients and healthy individuals.Compared to the traditional experimental methods,bioinformatics can explore the hidden molecular mechanisms of diseases,and is regarded as a highly effective research method.A study has discovered a shared biological connection between T2DM and AD through bioinformatics analysis.This connection is strongly associated with synaptic vesicle function and the MAPK signaling pathway[4].Additionally,the immune system has been found to play a significant role in this link[5].In a recent investigation,it was discovered that both AD and Metabolic syndrome exhibit the presence of immune cell infiltrations[6].Furthermore,the shared genes implicated in numerous metabolic pathways are closely linked to diverse immune cells.However,there are few studies on diabetic cognitive impairment using the hippocampus for bioinformatics analysis.There are no bioinformatics studies about immune-related genes and diabetic genes analyzed in cognitive impairment.Bioinformatics approaches may provide us with novel molecules associated with diabetic cognitive impairment that have not been fully studied.
Our study employed advanced bioinformatic methods to conduct a comprehensive analysis of immune-related genes,aiming to elucidate the regulatory mechanisms of these genes on cognitive function in individuals with T2DM and explore their underlying pathogenic pathways.Through bioinformatics,we identified differentially expressed genes (DEGs) in the hippocampus of diabetic and normal control mice.Meanwhile,we identified module genes most strongly associated with T2DM.We examined the role of immune-related genes in the progression of cognitive impairment linked to T2DM by conducting functional enrichment analysis,protein-protein interaction (PPI) analysis,and analysis of immune cell infiltration.Afterward,machine learning was utilized to identify three hub genes.We also identified the potential drugs associated with hub genes.In the end,we confirmed the expression of these three genes in mice and BV2 cells.Through these insights,we aspire to provide valuable information for the development of more efficacious treatment strategies and the enhancement of the quality of life of patients with T2DM-related cognitive impairment.
MATERlALS AND METHODS
Data collection and processing
The Gene Expression Omnibus (GEO) database[7] provided the datasets GSE125387 and GSE152539,which are associated with T2DM.The dataset GSE125387 consists of high-throughput sequencing data obtained from the hippocampus tissues of db/db mice (a mouse model for T2DM;n=10) and db/m mice (control mice;n=11).The Morris Water Maze test has validated distinct cognitive abilities in the two groups of mice,indicating that db/db mice exhibit deficiencies in learning and memory[8].GSE152539 consists of microarray expression data from hippocampus tissues of mice with high-fat dietfed (HFD) for 12 months (diabetic model mice;n=3) and mice with normal control diet-fed (n=3)[9].GSE125387 served as the primary dataset for analysis,while GSE152539 was utilized for hub gene validation.
Genes associated with immunity were acquired from Immunology Database and Analysis Porta (ImmPort)[10] and Mouse Genome Informatics (MGI)[11].ImmPort can be accessed at https://www.immport.org.and MGI at http://www.infor-matics.jax.org.
Analysis of differential expression genes
With GSE125387,we performed DEGs analysis by converting FPKM data into TPM data.The analysis for Principal Component Analysis (PCA) was conducted using the R software package called "stats"[12].To identify DEGs between the experimental group (db/db) and control group (db/m),we utilized the R package "limma" for differential analysis[13].In particular,we used |FoldChange| > 1.2 and FDR < 0.05 as filtering criteria.A volcano plot was generated using the R package "ggplot2"[14],and a heat map plot was created using the "ComplexHeatmap" package[15].
Functional enrichment analysis
To conduct the functional enrichment analysis on DEGs,we utilized Metascape (metascape.org/)[16].The analysis covered different platforms like Reactome[17],Gene Ontology (GO)[18,19],Kyoto Encyclopedia of Genes and Genomes (KEGG)[20],and Wiki pathways[21].The focus of the analysis was on DEGs,module genes,and immune-related DEGs.
The examination of potential biological functions and pathways in the hippocampus for both T2DM and normal groups within the predefined gene set was conducted using Gene Set Enrichment Analysis (GSEA)[22].All the genes were subject to analysis.The gene sets identified as "M2 curated gene sets" were obtained from the MSigDB database at https://www.gsea-msigdb.org/gsea/msigdb/collections.jsp.GSEA was implemented using the "clusterProfiler" package[23].
Weighted gene co-expression network analysis
Initially,we computed the deviation of every gene utilizing the gene expression patterns from GSE125387 and eliminated the lowest 75% of genes.To eliminate outliers in genes and samples,the goodSamplesGenes method from the R package "WGCNA" was employed[24].Afterward,we built a co-expression network with a scale-free property.The co-expression network was built using various criteria,which involved employing a soft thresholding function set at a power of 3.This function followed the scale-free topology criterion and yielded an independent index with anR2value of 0.85.Additionally,a minimum of 50 genes were required for each module in conjunction with the dynamic tree-cut method used for module merging,and a threshold of 0.5 was established.We set the sensitivity to 2,ultimately yielding 14 coexpression modules.Further analysis involved the use of Pearson correlation to examine any potential correlation between modules and groups.Moreover,an analysis of functional enrichment was conducted,as previously described.
Identification of immune-related DEGs
To identify immune-related DEGs,we cross-analyzed DEGs,key module genes found through Weighted Gene Co-Expression Network Analysis (WGCNA),and immune-related genes.The overlapping genes were visually presented using the R package "VennDiagram"[25].Moreover,an analysis of functional enrichment was conducted,as previously described.
Protein-protein interaction network construction
In order to examine the connections among protein-coding genes,we used the STRING database (string-db.org/)[26],with a specified minimum interaction score of 0.400.Nodes obtained from STRING were subsequently modified using Cytoscape software (3.9.1),and key interacting genes were identified with the aid of the CytoHubba plugin.The top 15 genes were independently ranked by Maximal Clique Centrality (MCC),Density of Maximum Neighborhood Component (DMNC) and Maximum Neighborhood Component (MNC).
Machine learning
For diagnosis,two additional machine learning algorithms were employed to further screen candidate genes.The technique of Lasso regression was utilized for variable selection and regularization,thereby enhancing predictive accuracy[27].For this research,we employed the R software package "glmnet" to perform regression analysis with the Lasso technique[28].In addition,we established a 3-fold cross-validation in order to acquire the most suitable model.The value of Lambda was adjusted to 0.05.In the meantime,the Random Forest (RF) algorithm was utilized due to its lack of limitations on variable conditions and ability to offer improved accuracy,sensitivity,and specificity[29].The RF analyses were conducted through the R package "randomForest"[30].Further diagnosis involved considering the hub genes obtained from the combination of Lasso and RF cross genes.
Receiver operating characteristic evaluation
Receiver operating characteristic (ROC) curve analysis was utilized to evaluate the diagnostic and discriminative significance of immune-related genes in cognitive impairment associated with T2DM.In order to measure the diagnostic worth,we computed the area under the curve (AUC) and assessed its significance by determining the 95% confidence interval (95%CI).The data was analyzed using the R package "pROC" to conduct ROC analysis[31].The GSE152539 dataset was employed as the external validation dataset.
Immune cell infiltration analysis
The analysis of tissue gene expression profiles using CIBERSORT,a computational technique,allows for the determination of the quantity of various immune cells[32].The immune infiltration of GSE125387 data was computed in this investigation by utilizing the markers of 25 immune cells in mice[33].For the analysis of immune cell infiltration,we utilized the dataset of mouse immune genes as the gene feature[33] through the CIBERSORTx website (https://cibersortx.stanford.edu/index.php/).We provided Supplementary Table 1 with the markers of 25 immune cells as the signature matrix.Bars were used to represent the distribution of immune cells in various samples.Comparisons between the proportions of various immune cell types in the diabetic and control groups were made using violin plots.To illustrate the connection between the hub genes and immune cells,we conducted a pairwise correlation analysis using the "Spearman" method.The outcomes of the analysis were then presented through heat maps utilizing the R package "ggplot2"[14].
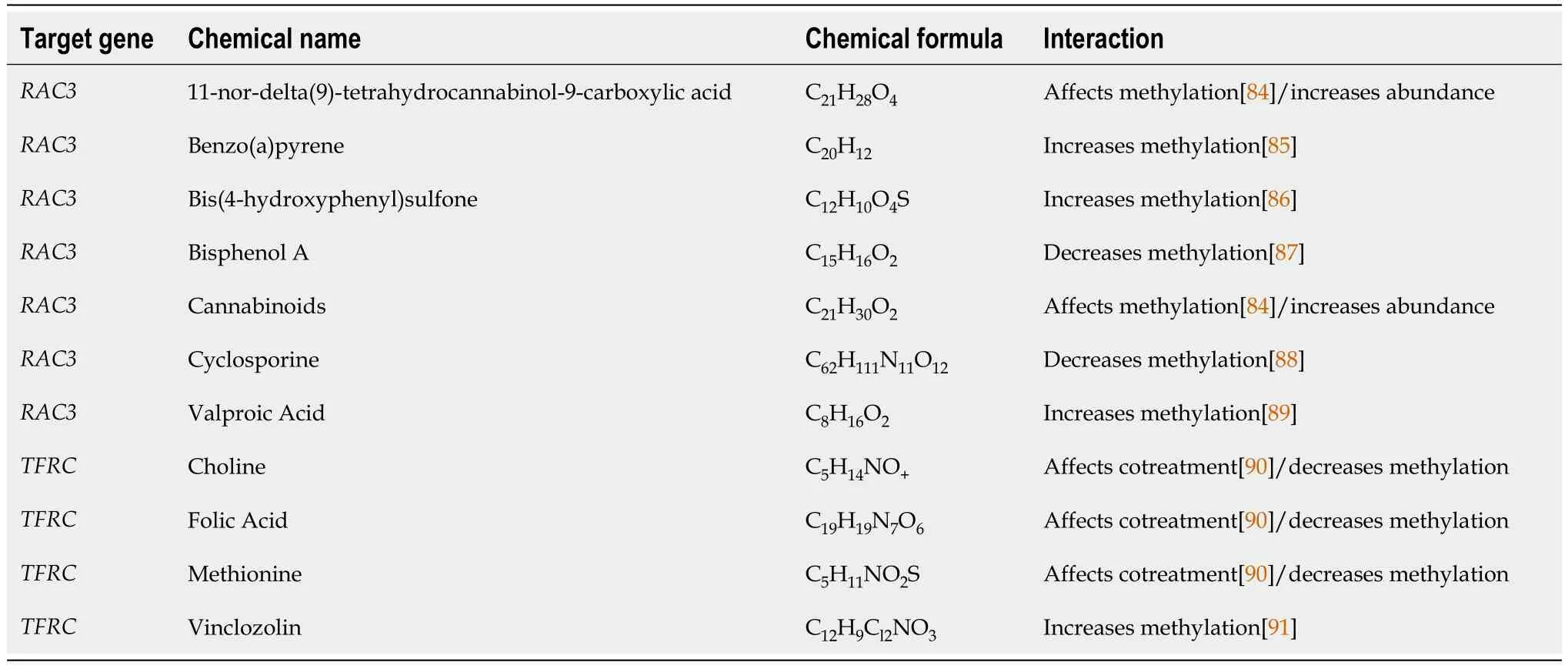
Table 1 Eleven potential compounds were selected by Comparative Toxicogenomics Database
Potential drug analysis
This study aims to analyze potential drugs that are effective in treating cognitive impairment in individuals with T2DM.Rac3andTfrcwere employed to identify potentially efficacious medications for Comparative Toxicogenomics Database (CTD) (https://ctdbase.org/) correspondingly.Afterward,we utilized PubChem (https://pubchem.ncbi.nlm.nih.gov/) to obtain the molecular formulas and two-dimensional structures of potential medications,aiding in the investigation of drugs.
Experimental animals and ethics
Male BKS.Cg-Dock7m+/+Leprdb/J homozygous Leprdb/dbmice were diabetic,and heterozygous Leprdb/mmice were used as controls (denoted as db/db and db/m in the text) in this study.As a diabetic model,the mice we used were consistent with the dataset GSE125387.A total of 9 male db/db mice aged six weeks and 9 male db/m mice aged six weeks were acquired from Jiangsu Hhuachuang sinoPharmaTechCo.,Ltd.Mice were raised in the Animal Research Center of Shandong University and housed in an environment of SPF level standards.The mice were kept in a chamber where the temperature was regulated within the range of 22°C to 25°C.The humidity is regulated within the range of 50% to 60% and the environment follows a 12-h cycle alternating between light and darkness.Autoclaving water was used for drinking and food was taken ad libitum.After being fed a regular diet for 24 wk,the mice were euthanized in order to collect brain tissue.The bilateral hippocampus of 3 mice from each group randomly selected was isolated for total RNA and protein extraction,respectively.While the cerebral hemispheres of remaining mice from each group were made into paraffin sections.
The Experimental Animal Ethics Review Committee of Qilu Medical College of Shandong University granted approval for our animal protocol (No.23001).The study adhered to principles that support the protection,well-being,and ethical treatment of animals,and it also complied with applicable national regulations concerning the welfare of laboratory animals.
Cell line culture and treatment
The BV-2 cell line (mice microglia) was cultured in DMEM/high-glucose medium (Gibco,United States) supplemented with 10% fetal bovine serum (Gibco,United States),1% penicillin,and streptomycin.The cells were incubated at 37°C with 5% CO2.Palmitic acid (PA) is a common saturated fatty acid.It is the main component of HFD and it has been found increased in the circulation of obese and diabetic people.PA has been studied in various biological contexts including inflammation,metabolic disorders,and cell signaling.In the central nervous system,PA has been associated with inflammatory responses.PA is recognized as a T2DM modelinvitro,such as in BV2 cells[34],β cells[35],and skeletal muscle cells[36].Changes in the cerebral gene expression profiles seemed to be specific in the T2DM model,as no such alterations were found in the type 1 diabetes mellitus model[37].So we chose the high-fat model instead of the high-glucose model.BV-2 cells were treated with 0.4 mmol/L PA (Sigma-Aldrich,United States) for a duration of 24 h,followed by extraction of total RNA or protein.
Reverse transcription quantitative real-time PCR
Total RNA was extracted from hippocampus or cells using the Total RNA Isolation Kit (Vazyme RC101) following the provided instructions.Reverse transcription was then performed with a Reverse Transcription Kit (Vazyme R323-01) and Thermal Cycler (Life Technologies,2720).Q-PCR was done with SYBR Green (Vazyme Q711-02/03) and Quantitative real-time PCR system (Roche,LightCycler480).The primer sequences were created and compared using the Primer-BLAST website (ncbi.nlm.nih.gov/tools/primer-blast/index.cgi?LINK_LOC=BlastHome),and subsequently synthesized by Beijing Tsingke Biotech Co.,Ltd.The expression of the target gene,relative to the β-Actin gene,was represented as 2ΔΔCt.
Western blotting
The samples were extracted from hippocampus tissue or cells and subsequently boiled in a loading buffer for 10 min.Protein separation was accomplished through SDS-PAGE (Epizyme,PG113).Primary antibodies,such as β-TUBLIN (Dilution 1:10000,Abways AB0039),RAC3 (Dilution 1:2000,Abcam ab129062),and TFRC (Dilution 1:1000,BOSTER PB9233),were incubated with the PVDF membranes overnight at 4°C.Afterward,the membranes were exposed to a secondary antibody (Dilution 1:10000,ZSGB-BIO ZB2305,ZB2301) for a duration of 2 h at room temperature.Subsequently,the electrochemiluminescence system was employed to detect the presence.
Immunohistochemistry
The paraffin sections were dewaxed,rehydrated,and subjected to antigen retrieval.To deactivate the natural peroxidase,a solution of 3% hydrogen peroxide was applied for a duration of 15 minutes.Following a 1-hour treatment with 5% BSA (bovine serum albumin),the slices were incubated overnight with primary antibodies.These antibodies included RAC3 (Dilution 1:100,Abcam ab129062) and TFRC (Dilution 1:100,BOSTER PB9233).On the next day,the paraffin sections were incubated with a secondary antibody (Genetech GK600505) at room temperature for a duration of 2 h.In the end,the slides underwent staining with a DAB Detection Kit (Genetech GK600505) and were subsequently counterstained with hematoxylin.
Data analysis
Three independent experiments were conducted and the data were presented as the mean ± SEM.Confirmation of data normality was established through the utilization of the Shapiro-Wilk test.To assess the distinction between two groups,Student's t-test was employed for data that followed a normal distribution.The Wilcoxon rank sum test was employed for data that did not follow a normal distribution.A statistically significant difference was defined as aPvalue < 0.05.Data analysis was performed with the use of R software (4.2.3) and Prism 9.
RESULTS
The Process of PCA and detection of DEGs
The flow chart for this study is illustrated in Figure 1.Using GSE125387,we identified DEGs between T2DM and control mice.Meanwhile,we identified module genes most strongly associated with T2DM using WGCNA.All the genes were subject to GSEA for functional enrichment analysis.By taking the intersection of DEGs,T2DM module genes,and immune-related genes,a total of 59 genes associated with the immune system were identified.Afterward,PPI and machine learning (Lasso regression and RF) were utilized to identify three hub genes (H2-T24,Rac3,andTfrc).Immune cell infiltration analysis was performed.The three hub genes were validated in GSE152539.Validation experiments were conducted at the mRNA and protein levels bothinvivoandinvitro.Additionally,11 potential drugs associated withRAC3andTFRCwere identified based on CTD.

Figure 1 Flow chart of the methods used in this study. DEGs: Differentially expressed genes;WGCNA: Weighted Gene Co-Expression Network Analysis;GSEA: Gene Set Enrichment Analysis;PPI: Protein-protein interaction;ROC: Receiver operating characteristic;RT-qPCR: Reverse transcription quantitative realtime PCR;WB: Western blotting;IHC: Immunohistochemistry.
The PCA indicated that the db/db group and db/m group were distinctly separated into two separate groups (Figure 2A).A total of 576 DEGs were detected,consisting of 214 genes showing upregulation and 362 genes exhibiting downregulation (Figure 2B).A heatmap displayed the most significant DEGs (Figure 2C).

Figure 2 Differentially expressed genes in GSE125387 and functional enrichment analysis. A: Principal component analysis shows that the db/db group and db/m group were distinctly separated;B: The volcano plot illustrates the distributions of differentially expressed genes (DEGs),with 214 genes showing upregulation (represented by red dots) and 362 genes showing downregulation (represented by blue dots).No significantly changed genes are marked as black dots;C: Heat map plot of the most significant DEGs;D: Analysis of functional enrichment for DEGs;E: Gene Set Enrichment Analysis (GSEA) between all the genes showing the up-regulated pathways;F: GSEA between all the genes showing the down-regulated pathways.PCA: Principal component analysis.
ImmPort and MGI provided a collection of 4142 genes related to mouse immune system.
Analysis of functional enrichment
Functional enrichment analysis was conducted using DEGs between db/db group and db/m group.The results demonstrated that DEGs were enriched in the "Microglia pathogen phagocytosis pathway" (WikiPathways);"positive regulation of endocytosis","circulatory system process","positive regulation of immune response","negative regulation of cell population proliferation","immune effector process" and "behavioral response to ethanol"(GO);"platelet activation,signaling and aggregation" and "metabolism of amine-derived hormones" (Reactome);"VEGF signaling pathway -Mus musculus (house mouse) " (KEGG;Figure 2D).GSEA results revealed that,in comparison to the db/m control group,"overlap between signal transduction pathways contributing to LMNA laminopathies" and "iron uptake and transport" pathways were upregulated in the db/db group (Figure 2E);"neuroactive ligand-receptor interaction" and "collagens" were downregulated (Figure 2F).The detailed enrichment items are listed in Supplementary Table 2 and 3.
Weighted gene co-expression network analysis
To approximate the scale-free structure of the network,we utilized a soft thresholding value of 3 (Figure 3A and B).We combined the top 25% of genes with the greatest variability into 14 co-expression modules by means of clustering (Figure 3C).Next,we conducted a Pearson correlation analysis to explore the connections between genes that define the modules and the traits of the groups.Our findings revealed that the Brown Module,comprising 974 genes,exhibited a significant association with the "group" trait (db/db and db/m) and displayed the strongest correlation (Figure 3D and E).In addition,we carried out functional enrichment analysis for the genes within the Brown Module.It showed the genes were enriched in "Collagen chain trimerization" and "Neuronal System" (Reactome);"behavior","regulation of membrane potential","synaptic signaling" and "locomotory behavior" (GO);"Serotonin and anxiety-related events" (WikiPathways;Figure 3F,Supplementary Table 4).
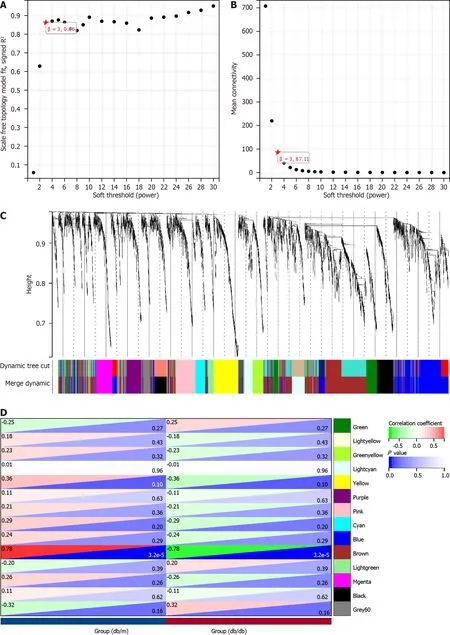
Figure 3 ldentification of module genes via Weighted Gene Co-Expression Network Analysis. A and B: The soft threshold β=3 is chosen as the result of the combined analysis of scale independence and average connectivity;C: Different colors represent gene co-expression modules in the gene tree;D: Heatmap illustrating the correlation between modules and type 2 diabetes mellitus (T2DM).There is a significant correlation between the brown module and T2DM;E: The brown module contains 974 genes,and the scatter diagram shows the correlation between membership in the brown module and gene significance for the group;F: Analysis of gene enrichment in the brown module for functional purposes.
Identification of Immune-related DEGs
The cognitive impairment of T2DM mice (db/db) that we utilized has been confirmed[8].The above analysis of enrichment indicated a strong association between DEGs and the immune system.In order to explore the connection between cognitive impairment related to diabetes and immunity,we utilized a Venn diagram (Figure 4A) to identify 59 genes that overlapped between DEGs,genes in the Brown Module,and immune-related genes.Figure 4B displays the heat map of the 59 genes.The analysis of gene function enrichment revealed that the immune-related DEGs were highly concentrated in pathways such as "Microglia pathogen phagocytosis pathway" (WikiPathways),"positive regulation of immune response","regulation of leukocyte cell-cell adhesion","regulation of neuron death",and "regulation of behavior".Additionally,the pathways "Axon guidance -Mus musculus (house mouse) " and "B cell receptor signaling pathway -Mus musculus (house mouse)" (KEGG) as well as "ER-Phagosome -Mus musculus (house mouse)" (Reactome) were also enriched (Figure 4C and Supplementary Table 5).Summary of enrichment analysis in PaGenBase about the prediction of specific cell types for the 59 immune-related DEGs showed they were predicted in microglia (Figure 4D).

Figure 4 ldentification of immune-related differentially expressed genes and functional enrichment analysis. A: The intersection of differentially expressed genes (DEGs),immune-related genes,and brown module genes via Weighted Gene Co-Expression Network Analysis reveals a total of 59 identified genes;B: Heat map plot of the 59 immune-related DEGs;C: Functional enrichment analysis was performed on the 59 genes;D: Prediction of specific cell types for the 59 genes.DEGs: Differentially expressed genes.
PPI network
The PPI network is shown in Figure 5A,with 35 genes displaying interaction capabilities.The visualization was performed utilizing the CytoHubba plugin within Cytoscape (Figure 5B).The MCC,DMNC,and MNC methods independently ranked the top 15 genes.The significance of the interaction network increases as the color becomes darker (Figure 5C-E).The overlap of the top 15 genes acquired through the three approaches resulted in 11 genes (Figure 5F).

Figure 5 Construction of a network for the interaction between proteins. A and B: The protein-protein interaction network analysis shows that 35 genes have interactions with one another;C-E: Top 15 genes were independently ranked by Maximal Clique Centrality,Density of Maximum Neighborhood Component and Maximum Neighborhood Component methods.The scores were ordered by color from red to yellow;F: The overlap of the top 15 genes obtained from the three methods resulted in a total of 11 genes.MCC: Maximal Clique Centrality;DMNC: Density of Maximum Neighborhood Component;MNC: Maximum Neighborhood Component.
Identification of hub genes using machine learning
To evaluate the diagnostic significance of potential genes,we utilized Lasso regression and RF machine learning techniques.Figure 6A and B revealed that Lasso regression detected 4 possible biomarker contenders namelyC1qa,H2-T24,Rac3,andTfrc.The genes were ranked according to their importance by the RF algorithm (Figure 6C and D).To depict the overlap between the 4 possible contenders in Lasso and the leading 5 genes in RF,a Venn diagram was employed,leading to the identification of 3 genes (H2-T24,Rac3,andTfrc) for the ultimate validation phase (Figure 6E).
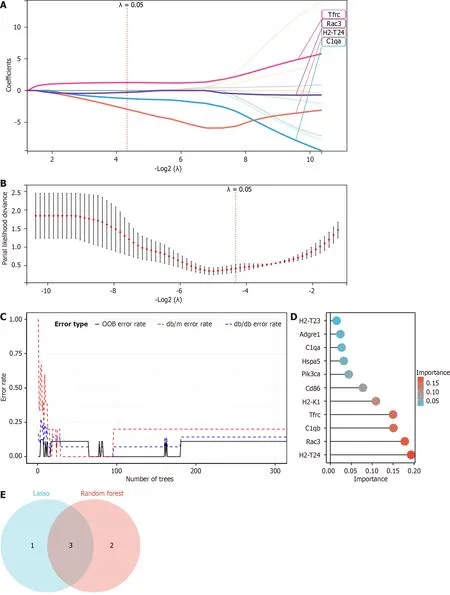
Figure 6 Utilizing machine learning to evaluate potential diagnostic biomarkers in candidate screening. A and B: Screening of biomarkers in the Lasso model.For diagnosis,the most appropriate gene count (n=4) is the one that corresponds to the lowest point on the curve;C and D: The error is displayed by the random forest algorithm.The importance score is used to rank genes;E: The Venn diagram illustrates that the above two algorithms have identified three potential diagnostic genes: H2-T24,Rac3 and Tfrc.
ROC evaluation
In GSE125387,the findings were as follows:H2-T24(AUC 1.000,95%CI: 1.000-1.000),Rac3(AUC 0.982,95%CI: 0.939-1.000),andTfrc(AUC 0.991,95%CI: 0.966-1.000,Figure 7A-C).Figure 7D displayed the expression levels of these three genes in GSE125387.The GSE152539 dataset was employed as the external validation dataset.The findings were as follows:H2-T24(AUC 0.667,95% CI 0.013-1.000),Rac3(AUC 1.000,95%CI: 1.000-1.000),andTfrc(AUC 1.000,95%CI: 1.000-1.000,Figure 7E-G).In GSE152539,the expression levels of three genes were also confirmed (Figure 7H).

Figure 7 Evaluation of the receiver operating characteristic. A-C: In GSE125387,the receiver operating characteristic (ROC) curve for each candidate gene (H2-T24,Rac3,and Tfrc) was analyzed;D: The levels of expression for the three genes in GSE 125387;E-G: In GSE152539,the ROC curve for each candidate gene (H2-T24,Rac3,and Tfrc) is displayed;H: The levels of expression for the three genes in GSE152539 were also confirmed.aP < 0.05.AUC: Area under the curve;95%CI: 95% confidence interval;TPR: True-positive rate;FPR: False-positive rate.
Immune cell infiltration analysis
To gain a deeper understanding of the immune regulation involved in the hippocampus of T2DM,we performed an analysis of the infiltration of immune cells.We utilized 25 mouse immune cells as feature genes to determine the relative abundance of each immune cell in the samples from the db/db and db/m groups.The results were visualized in a bar graph (Figure 8A).According to the violin plot,the level of "M2 Macrophage" (P=0.082) was higher in the db/db group compared to the db/m group.Conversely,the levels of "T Cells CD4 Follicular" (P=0.051) and "M0 Macrophage" (P=0.089) were lower in the db/db group.There is a tendency of effect but it is not statistical.The db/db and db/m groups did not show expression of "T Cells CD8 Actived","Treg Cells",and "T Cells CD4 Memory" (Figure 8B).Moreover,the correlation heatmap exhibited associations among various immune cell types and 3 hub genes.We observed the infiltration of various immune cells in diabetic mice.The strongest synergistic effect was observed between the categories of "T Cells CD4 Follicular" and "M0 Macrophage" (r=0.841),with the subsequent highest correlation found between "B Cells Naive" and "M0 Macrophage" (r=0.709).On the other hand,the most significant competitive impact was observed between "NK Resting" and "NK Actived" (r=-0.796),with "B Cells Naive" and "B Cells Memory" (r=-0.795).A variety of immune cells were linked toH2-T24,Rac3,andTfrc.The study's correlation analysis revealed a negative association betweenH2-T24and "Th2 Cells".The presence ofRac3showed a positive correlation with "M0 macrophage".The association ofTfrcwith "Neutrophil Cells","M0 Macrophage",and "T cells CD4 Follicular" was negative,whereas it was positive with "T cells CD4 Naive"(Figure 8C).

Figure 8 lmmune cell infiltration analysis to gain a deeper understanding of the immune regulation involved in the hippocampus of type 2 diabetes mellitus. A: The bar plot visualizes the distribution of 25 different types of immune cells across various samples;B: The violin plot visualizes the comparison of the proportion of 25 types of immune cells between db/db and db/m;C: The correlation between the compositions of 22 types of immune cells and 3 hub genes.Various immune cells show the infiltration.Cor: Correlation coefficient.
Identifying potential drugs
To illuminate the individualized therapy for cognitive impairment in diabetes,researchers identified small molecule drugs targetingRac3andTfrc.11 associated drugs were identified (Table 1,Figure 9).These drugs mainly affect themethylation of genes or promoters.It is worth noting thatH2-T24is not a homologous gene in humans.Therefore,we decided not to proceed with the drug predictions that were planned.

Figure 9 Two-dimensional structures of potential compounds. A: 11-nor-delta(9)-tetrahydro;B: Benzo(a)pyrene;C: Bis(4-hydroxyphenyl)sulf;D: Bisphenol A;E: Cannabinoids;F: Cyclosporine;G: Valproic Acid;H: Choline;I: Folic Acid;J: Methionine;K: Vinclozolin.
Validation of the expression of hub genes in vivo and in vitro
Supplementary Table 6 displays the primer sequences.Student's t-test was employed for data.The mRNA expression of 3 hub genes (H2-T24,Rac3,andTfrc) was validated with reverse transcription quantitative real-time PCR (RT-qPCR).In comparison to db/m mice,H2-T24andRac3showed a reduction in expression in db/db mice (P< 0.01;P< 0.05),whereasTfrcdemonstrated an elevation in expression in db/db mice (P< 0.01;Figure 10A).PA is a common saturated fatty acid.PA was used as a T2DM modelinvitro.Similar to the findings in mice experiments,H2-T24andRac3showed a reduction in expression in the PA group (P< 0.05),whereasTfrcdemonstrated an elevation in expression in the PA group (P< 0.05;Figure 10B).Subsequently,we conducted additional verification of the protein levels of RAC3 and TFRC throughinvivoandinvitrowestern blotting.The findings indicated that the protein levels of RAC3 and TFRC were in agreement with the mRNA levels (P< 0.05) as demonstrated in Figure 10C-F.In the hippocampus of db/db mice,immunohistochemistry revealed a decrease in RAC3 expression and an increase in TFRC expression,as compared to db/m mice (Figure 10G).
DISCUSSION
The worldwide incidence of T2DM is on the rise,occurring concomitantly with target organ damage and poor prognosis,often linked to the occurrence and development of cognitive impairment.Several mechanisms have been proposed to explain the relationship between T2DM and cognitive impairment,including hyperglycemia,insulin resistance,vascular impairment,oxidative stress,and neuroinflammation[3].Neuroinflammation is a term used to describe the stimulation of glial cells,specifically microglia and astrocytes,leading to the generation of inflammatory cytokines and chemokines within the central nervous system[38].A study found that knockout of TLR2 protected against diabetes-induced cognitive impairment[39].Similarly,suppression of NLRP3 enhances cognitive ability and maintains vascular health following stroke in diabetic animals[40].Hence,it is imperative to conduct additional research to investigate the causes of cognitive impairment in diabetes and discover possible treatment targets.
In this study,several biological information research methods were used to obtain DEGs related to T2DM in mice from GEO high-throughput sequencing datasets.Pathway enrichment analysis showed that DEGs were linked to microglia pathogen phagocytosis,immune inflammation,and collagen synthesis.Our study focused on analyzing the interaction between immune regulation and the development of cognitive impairment in diabetes to explore relevant targets.The results of this research may enhance comprehension regarding immune impairment,neuroinflammatory processes,and their impact on cognitive impairment in individuals with diabetes.
Currently,immune-related genes associated with cognitive impairment have been identified,but there is a lack of bioinformatics studies on diabetic cognitive impairment.In this study,we utilized the immune databases from MGI and ImmPort to identify immune-related DEGs.To further characterize diabetes phenotype genes,we performed a WGCNA analysis.We conducted a cross-analysis of DEGs,key module genes found through WGCNA,and immune-related genes to identify DEGs associated with the immune system.Our research found that immune-related DEGs were highly concentrated in the immune response and phagocytosis pathways.The summary of the enrichment analysis showed that immune-related DEGs were predicted in microglia.
An increasing number of evidence suggests a connection between microglia and cognitive decline.Microglia are the main components of the brain's natural defense system,and play a vital role in neuroinflammation,which is strongly linked to cognitive impairment associated with T2DM[41,42].Activation of microglia can lead to neuroinflammation and neuronal damage,resulting in cognitive impairment[43].A review highlighted the complex role of microglia in cognition,including their involvement in neurogenesis,synaptic pruning,and learning and memory.And it may hold potential targets for treating cognitive impairment[44].A single-cell sequencing study confirmed the opinion that microglia in the hippocampus and immune system play a vital role in diabetes-associated cognitive impairment[45].Focusing on the immune system and neuroinflammation could offer a promising pathway for creating new treatment approaches to enhance cognitive abilities in T2DM.
Through machine learning,finally,3 immune-related DEGs have been identified.To verify our findings,we established a mouse model of T2DM and performed quantitative analysis of gene expression,which revealed that 3 genes -H2-T24,Rac3,andTfrc -showed expression trends consistent with our bioinformatics results.
H2-T24(Histocompatibility 2,T region locus 24) has been shown to play an important role in immune function.H2-T24is associated with the microglia activation after cerebral ischemia[46].H2-T24has a significant impact on the development of AD[47].H2-T24is associated with hippocampus-based memory impairment by endogenous retrovirus[48].These findings suggest thatH2-T24is related to neuroinflammation.However,research onH2-T24in cognitive impairment is limited.Until now,research on the association betweenH2-T24and diabetes is rare.However,it is worth noting thatH2-T24is not a homologous gene in humans.Therefore,we decided not to proceed with the drug predictions that were planned.
Rac3,a small GTPase belonging to the Rho family,is primarily found in the brain[49].Rho family GTPase signaling pathways have been proposed to be linked to diabetes[50].Racfamily can inhibit PTEN[51],which is a critical negative regulator in the PI3K pathway of insulin signaling[52,53].We propose that theRacfamily has an effect on insulin signaling through inhibiting PTEN,andRac3deficiency affects glucose homeostasis and insulin sensitivity.We consider that insulin resistance is a contributing factor to cognitive dysfunction.Rac3plays a critical role in the regulation of dendritic spine development and synaptic plasticity in the hippocampus[54].The activation of STAT3 and ERK byRac3stimulates the proliferation and migration of glioma cells[55].Rac3is crucial for regulating microglial activation and neuronal inflammation in response to brain injury related to the HMGB1 signaling pathway[56].In an AD study,the expression ofRAC3was decreased[57].Our study's dataset and experimental validation revealed that the expression ofRac3was notably reduced in T2DM mice,potentially resulting in cognitive decline.Further research on its functions and potential therapeutical applications in neurodegenerative disorders is needed.
The expression ofTrfc(transferrin receptor) is significantly increased in neural tissues during neuronal regeneration and repair[58].The transport of iron across the blood-brain barrier,which is crucial for appropriate neuronal activity,is controlled byTrfc[59].Recent studies have linkedTrfcto neuroinflammatory conditions.Tfrcwas upregulated in activated microglia in rats with central pain[60].Tfrcwas significantly upregulated in the brains of Parkinson's disease[61].In AD,Trfcis overexpressed,leading to altered iron homeostasis and oxidative stress[62].The reduction of iron in the brainpresents a new approach to treating AD,indicating thatTfrcmay serve as a promising target for therapeutic intervention in AD[63].In another AD study,the expression ofTFRCwas increased[57].However,there are some conflicting conclusions aboutTrfcin AD.For instance,patients with AD experience a notable reduction inTfrclevels within their peripheral blood mononuclear cells[64].Hence,further comprehensive and thorough investigations are required to explore the function ofTfrcin the nervous system.In another bioinformatics analysis study,Tfrchas been described as playing a role in T2DM and neurological diseases[65].In T2DM,increased iron stores have been found to predict the development of the disease[66].CirculatingTfrcis associated with the relationship among post-load glucose,insulin resistance,and T2DM[67].We consider thatTfrcoverexpressed is associated with insulin resistance and thus contributes to cognitive dysfunction.Additional investigation is required to clarify the correlation betweenTfrcand neuroinflammation,as well as to examine the possibility ofTfrcas a target for therapy.
Metabolism and the immunological state are unavoidably intertwined[68].The metabolic disorders,such as hyperglycemia and hyperlipidemia,induce a state of inflammation in the body in individuals with T2DM.Immune dysregulation is common in cognition impairment.During the study,we employed CIBERSORT to examine the infiltration of immune cells and observed variations in several cell types between the diabetes group and the control group,despite the absence of any significant statistical disparity.This gives us an implication that different immune states affect cognitive function.
Eleven potential drugs were identified in this study.These drugs have a variety of functions,and when the dosage or duration of application is different,it is possible to have the opposite effects on cognitive function.Cyclosporine is an immunosuppressant,mainly used for rejection after organ and tissue transplantation.During surgery under general anesthesia,Cyclosporine treatment can increase ATP levels in the cerebral cortex and improve learning and memory function[69].Valproic acid is commonly used in the treatment of epilepsy and bipolar affective disorder.It is reported that it improves cognitive function in patients with bipolar affective disorder[70].However,long-term use of valproic acid can impair cognitive function[71].Valproic acid exposure can cause autism in prepubertal rats[72].Choline is a constituent of biological membranes and precursor of acetylcholine in cholinergic neurons.It can promote brain development and improve memory[73].Lifelong choline supplementation may ameliorate AD by attenuating microglial activation[74].It is well known that folic acid is closely related to fetal neurodevelopment.The deficiency of folic acid can result in elevated levels of homocysteine,thereby contributing to the development of atherosclerosis,stroke,diabetes,and other related conditions[75].Folic acid supplementation affects cognition and inflammation in patients with AD[76].A Methionine-restricted diet can improve cognitive function[77].Another study of Chinese adults revealed that animal methionine and plant methionine intake were positively and inversely associated with cognition[78].These drugs,which have beneficial effects on the nervous system,may become therapeutic options for diabetes-related cognitive impairment.However,11-nor-delta (9)-tetrahydrocannabinol-9-carboxylic acid[79],Cannabinoids[80],Benzo(a)pyrene[81],Bis(4-hydroxyphenyl)s,Bisphenol A[82] and Vinclozolin[83] are neurotoxic and can lead to cognitive impairment.This suggests that patients with diabetic cognitive impairment should avoid exposure to neurotoxic drugs.
Our study has certain restrictions.Firstly,given the scarcity of hippocampus datasets from diabetic mice with validated cognitive impairment,we used only one dataset for bioinformatics analysis and the other for validation of hub genes.Secondly,our validation of the hub genes has been limited to diabetic mice,and we do not have the backing of clinical data.Furthermore,despite performing a thorough bioinformatics analysis in this study,we did not carry out additional experiments to validate the impact of immune-related genes on cognitive function.Therefore,additional investigation is required to further explore the precise mechanism of immunometabolism regulation in diabetic cognitive impairment bothinvivoandinvitro.Our future investigation will concentrate on this new course.
CONCLUSlON
In summary,we identified the differences in immune-related genes in the hippocampus between T2DM and control mice by comprehensive bioinformatics analysis.The immune-related DEGs were closely related to microglia.3 hub genes were screened and verified-H2-T24,Rac3,andTfrc.They were associated with a variety of immune cells.We verified the expression of these 3 genesinvivoandinvitro,consistent with the bioinformatics analysis.11 drugs associated withRAC3andTFRCwere identified.These findings suggest that they are co-regulatory molecules of immunometabolism in diabetic cognitive impairment,and provide a new insight in the treatment of diabetic cognitive impairment.
ARTlCLE HlGHLlGHTS
Research background
Cognitive decline in type 2 diabetes mellitus (T2DM) is a complex and progressive condition that demands additional research for complete understanding.Neuroinflammation is seen as a primary mechanism,with the immune system significantly influencing the disease's advancement.
Research motivation
Cognitive impairment in T2DM is complex and evolving,necessitating deeper research.The immune system significantly impacts its progression.
Research objectives
To pinpoint and confirm hippocampus immune-related genes linked to cognitive impairment in T2DM.
Research methods
Using the Gene Expression Omnibus database GSE125387,we pinpointed genes differentially expressed between T2DM and control mice,and identified key module genes related to T2DM through Weighted Gene Co-Expression Network Analysis.We conducted Gene Set Enrichment Analysis for these genes and built a protein-protein interaction network,employing Lasso regression and Random Forest to identify three hub genes.These genes underwent immune cell infiltration analysis and were validated in GSE152539 using receiver operating characteristic curve analysis.Validation included RT-qPCR,Western blotting,and immunohistochemistry at mRNA and protein levels,bothinvivoandinvitro.Furthermore,we discovered 11 potential drugs linked to these genes using the Comparative Toxicogenomics Database.
Research results
We identified 576 DEGs from GSE125387 and intersected them with T2DM module and immune-related genes,finding 59 immune system-related genes.Machine learning pinpointed three hub genes (H2-T24,Rac3,Tfrc),linked to various immune cells.These genes were validated in GSE152539,with experiments at mRNA and protein levelsinvivoandin vitro,aligning with our bioinformatics analysis.Additionally,11 potential drugs related toRAC3andTFRCwere identified using the Comparative Toxicogenomics Database.
Research conclusions
The immune system plays a significant role in cognitive impairment in T2DM.The immune-related differently expressed genes in hippocampus were closely related to microglia.We confirmed the expression of three such genes bothinvivoandinvitro,in line with our bioinformatics findings.Three hub genes screened were associated with a variety of immune cells.Moreover,11 drugs related toRAC3andTFRCwere identified.
Research perspectives
These genes are as co-regulatory molecules in the immunometabolism of diabetic cognitive impairment,offering new perspectives for its treatment.
FOOTNOTES
Author contributions:The design of this study was carried out by Hou XG;the collection and analysis of bioinformatics data,experimental validation,and writing of the manuscript were carried out by Gao J;Zou Y took on the task of conducting statistical analysis on the experimental data;Lv XY was responsible for animal modeling;Chen L contributed to the literature research;the final manuscript was read and approved by all the authors.
Supported byNational Natural Science Foundation of China,No.82270845.
lnstitutional review board statement:All procedures involving animals were reviewed and approved by the Institutional Animal Care and Use Committee of the Qilu Medical College of Shandong University (IACUC protocol number: 23001).
lnstitutional animal care and use committee statement:All procedures involving animals were reviewed and approved by the Institutional Animal Care and Use Committee of the Qilu Medical College of Shandong University (IACUC protocol number: 23001).
Conflict-of-interest statement:The authors assert that there are no conflicting interests.
Data sharing statement:No additional data are available.
ARRlVE guidelines statement:The authors have read the ARRIVE Guidelines,and the manuscript was prepared and revised according to the ARRIVE Guidelines.
Open-Access:This article is an open-access article that was selected by an in-house editor and fully peer-reviewed by external reviewers.It is distributed in accordance with the Creative Commons Attribution NonCommercial (CC BY-NC 4.0) license,which permits others to distribute,remix,adapt,build upon this work non-commercially,and license their derivative works on different terms,provided the original work is properly cited and the use is non-commercial.See: https://creativecommons.org/Licenses/by-nc/4.0/
Country/Territory of origin:China
ORClD number:Xin-Guo Hou 0000-0003-2045-1290.
S-Editor:Lin C
L-Editor:A
P-Editor:Zhao S
 World Journal of Diabetes2024年4期
World Journal of Diabetes2024年4期
- World Journal of Diabetes的其它文章
- Metabolic syndrome’s new therapy: Supplement the gut microbiome
- Pancreatic surgery and tertiary pancreatitis services warrant provision for support from a specialist diabetes team
- Effect of bariatric surgery on metabolism in diabetes and obesity comorbidity: lnsight from recent research
- Application of three-dimensional speckle tracking technique in measuring left ventricular myocardial function in patients with diabetes
- lcariin accelerates bone regeneration by inducing osteogenesisangiogenesis coupling in rats with type 1 diabetes mellitus
- Long-term effects of gestational diabetes mellitus on the pancreas of female mouse offspring