
作物全基因组选择育种技术研究进展
2024-04-17 06:48王欣徐一亿徐扬徐辰武
生物技术通报 2024年3期
王欣 徐一亿 徐扬 徐辰武
(1.扬州大学农学院,扬州 225009;2.扬州大学信息工程学院,扬州 225009)
传统的作物育种基于表型选择,通过观察作物表型的变异选择优良后代。虽然育种家可以利用生物遗传一般规律、综合选择指数、同期群体比较和田间试验统计等手段进行田间试验设计和选择,但是其工作高度依赖于育种家的经验,效率较低。20世纪90 年代以来,伴随着基因组上大量分子标记的开发,人们开始借助分子标记进行辅助育种。
目前分子标记辅助选择育种技术的应用已经愈发成熟,但是其只适用于由较少主效QTL 决定的性状。实际的作物育种工作需要多个性状的协同改良,育种项目中可供育种家利用的材料有成百上千份,组配组合则更多,然而由于试验规模限制,大量重要材料并未进行测试,育种效率较低。全基因组选择(genomic selection,GS)[1]方法利用覆盖全基因组的分子标记和样本的表型数据建立预测模型,以实现个体的遗传评估。利用GS 技术开展育种工作,只需对较少的材料/品种进行表型鉴定,就可以利用基因组上的高密度标记对更多尚未开展田间试验的材料/品种表型进行预测,能够大大降低育种成本,提高育种效率。
GS 技术在动物育种尤其是奶牛育种中已经取得了很大进展,并且在加拿大、美国等国家的奶牛育种实践中得到了广泛应用。但是由于育种体系和育种目标的差异,作物的GS 面临若干不同的问题,如品种间缺乏明确的系谱关系,环境对表型有较大影响等。近年来随着高通量测序技术的发展和测序成本的下降,GS 技术在作物育种中也获得了较大发展。特别是作物的杂种育种中,杂交种的基因型可以由亲本基因型进行推断,GS 的优势更加突出。目前国内外已经开展了多种作物的GS 验证研究。如水稻中,Xu 等[2]从210 份重组自交系亲本所产生的21 945 份杂交后代中随机选择278 份材料进行表型鉴定,并利用这278 份材料作为训练样本来预测所有可能杂交种的产量相关性状,发现预测产量最高的100 个潜在杂交种的产量比平均产量提高16%。小麦中,Juliana 等[3]基于国际玉米和小麦改良中心(CIMMYT)48 562 个产量观测结果的大型数据集进行建模,在产量测试的第1、2 和3 阶段分别获得了0.56、0.50 和0.42 的平均预测精度。在热带玉米的多亲本育种群体中,Zhang 等[4]的研究指出,快速循环基因组选择是一种在短时间内既能保持遗传多样性又能获得高遗传增益的有效育种策略。
作为作物分子设计育种中一项不可或缺的先进技术,GS 是国际数量遗传学研究的重要热点,近年来在模型算法、群体构建方案、多性状与多环境预测方法和多组学预测方法等方面涌现出了大量研究成果。如Guo 等[5]深入研究了不同训练集设计方案对杂交种表型预测的影响,结果表明,对训练集的精心设计,能够显著提高模型的预测精度。Wang等[6]的研究将玉米亲本一般配合力(GCA)的估计和杂种表型预测相结合,提出了稀疏部分双列杂交(SPDC)设计方案,能够同时实现对玉米大量亲本GCA 值和更多杂交种表型的精确预测。Xu 等[7]在玉米中开展多组学联合分析的同时,整合双亲表型预测杂交种的表现,显著提升了表型预测的准确性。Yin 等[8]开发了运用机器学习确定模型参数的KAML 方法,并用于包括玉米、人类、牛和马的多个数据集,预测精度高于经典的GBLUP 和贝叶斯方法,展示出机器学习方法在GS 中的成功应用。近期Wang 等[9]开发了一种基于深度神经网络的GS方法DNNGP,其表现超过了GBLUP 和LightGBM 等多种经典方法。
理论方法的创新为GS 技术的发展奠定了基础,不过要在育种中落到实处,作物基因型和表型的高效测定是必要前提。近年来,单核苷酸多态性(SNP)标记在水稻、玉米、小麦和大豆等作物的资源鉴定、遗传分析、功能基因挖掘和分子设计育种等方面得到越来越广泛的应用。虽然芯片的SNP 标记密度低于重测序技术,但是其成本相对较低,准确度高,重复性好,试验流程标准化程度高,芯片设计灵活,为GS 中基因型数据的获取提供了重要技术条件。
GS 育种是分子设计育种的重要方法,自问世以来就享有“革命性育种技术”的美称。目前国外发达国家种业企业已经把作物GS 育种付诸实践,但是我国的作物GS 育种大多还处于实验室模拟阶段,其原因是多方面的,包括模型预测精度不够理想,基因型和表型数据共享程度低,缺少GS 育种专用芯片以及配套软件和平台等问题。本文旨在阐述并分析当前作物GS 的研究现状,指出其面临的问题和发展前景,为推动GS 的进一步发展提供策略和思路。
1 全基因组选择方法概述
GS 的实施过程,首先要采集训练群体的表型和基因型信息,然后利用模型估计各标记的效应,进而利用候选群体的基因型估计其遗传效应值[10]。然而在全基因组选择模型中,标记的数量P 往往大幅超过观测的样本数n,从而给模型的训练和目标性状的精确预测带来困难。近年来,大量学者开发出了一系列基因组选择方法,主要包括线性模型及其扩展,各类贝叶斯方法,以及多种机器学习(machine learning,ML)方法(图1)。
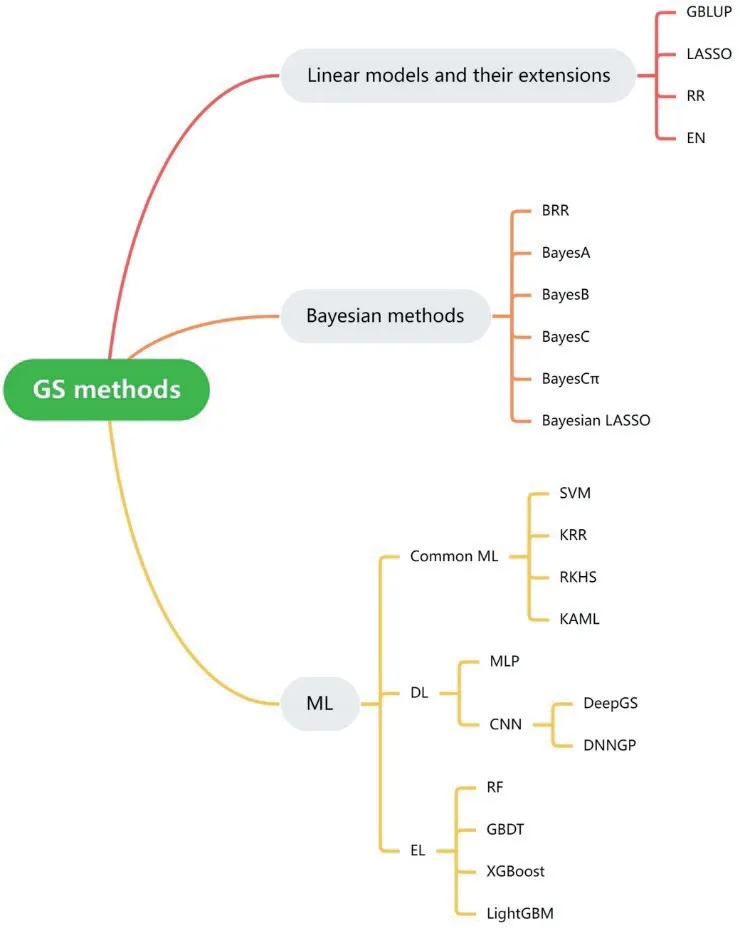
图1 GS 方法的分类Fig.1 Classification of GS methods
1.1 线性模型及其扩展
基因组最佳线性无偏预测(genomic best linear unbiased prediction,GBLUP)[11]是一种利用全基因组标记预测目标群体基因型值和表型值的高效方法[12-13]。它通过构建基因组关系矩阵G,代替传统BLUP 中基于系谱关系建立的亲缘关系矩阵。G 矩阵通常由全基因组上的高密度标记构建,而利用大量基因标记信息的最佳线性无偏估计则保证了GBLUP方法预测精度的稳健性,使其在广泛的各类数据集中都表现较好。此外,由于GBLUP 方法遗传效应的计算公式简单,且无需迭代运算,所以效率很高,是GS 研究中最常用的一般方法和各种创新方法的比较基准,也被大量学者用作复杂场景(如多组学、多性状和多环境研究)下GS 拓展方法的基础模型。最小绝对收缩和选择算子(least absolute shrinkage selection operator,LASSO)[14]在线性回归的基础上添加了L1 正则项,通过构造一个惩罚函数得到较为精炼的模型,将大部分标记的效应压缩为0,是一种选择收缩算法。Friedman 等[15]利用循环坐标下降法开发了快速求解LASSO 的算法,克服了大多数选择收缩算法耗时过长的问题,成为该方法的一大优势。岭回归(ridge regression,RR)在线性回归的基础上添加了L2 正则项[16],弹性网(elastic net,EN)[17]则同时使用L1 和L2 正则化,是LASSO 和岭回归的结合。
1.2 贝叶斯方法
贝叶斯类方法假定标记的效应服从一定的先验分布[18-19],其中BRR 假定所有标记的效应有着相同的方差;BayesA 则允许每个标记的效应有不同的方差;BayesB 和BayesC 中大部分的标记效应被设置为0,对于剩余少数标记的效应,BayesB 允许有不同的方差,BayesC 则假定它们有相同的方差。BayesCπ 在BayesC 的基础上,设定0 效应标记的占比为服从均匀分布的变量。从假设条件上看,BRR将各个标记均等对待,这一点与GBLUP 无差别利用大量标记信息计算G 矩阵的效果相似。BayesA 所估计得到的标记效应,差异稍大。BayesB、BayesC 和BayesCπ 则更进一步扩大了这种差异,其中BayesB几乎在最大程度上对标记效应进行选择收缩和差别对待,因此成为选择收缩算法的代表,特别适用于由少数主效基因决定的性状。各类贝叶斯方法具有设计精巧、准确性高和可解释性强等优点,但是模型的求解往往依赖于贝叶斯框架下的抽样方法求解,所以计算效率较低,给实际推广带来了一定的制约。
1.3 机器学习方法
机器学习的快速发展为GS 提供了更加丰富和灵活的方法。一般的机器学习模型包括支持向量机(support vector machine,SVM)[20]、再生核希尔伯特空 间(reproducing kernel Hilbert space,RKHS)[21]、核 岭 回 归(kernel ridge regression,KRR)[22]和KAML 等[8]。SVM 通过寻找最佳分隔的超平面,执行线性或非线性的分类和回归。RKHS 利用高斯核函数拟合模型,可通过贝叶斯框架下的抽样方法或混合线性模型求解。KRR 则在岭回归的基础上引入核函数,将原始空间中的数据映射到更高维的核空间,以实现对非线性函数的学习。
深度学习(deep learning,DL)[23]是机器学习的重要分支,它使用包含多个隐藏层的深度神经网络。相对于其他浅层结构算法,深度神经网络具有更强的特征学习能力,能够捕捉数据中蕴含的复杂非线性关系。尽管所有的深度学习方法都由多个神经元堆叠而成,但是它们实际上包括各种各样的架构,在GS 中应用较多的结构包括多层感知机(multilayer perceptron,MLP)和卷积神经网 络(convolutional neural networks,CNN)[24]。 如Montesinos‑López 等[25]曾利用7 组小麦数据集评估了MLP 的全基因组预测性能。同样基于MLP 结构,Montesinos‑López 等[26-27]利用关系矩阵的“克罗内克积”反映性状间和环境间的联系,进而实现了多性状和多环境的联合预测。CNN 技术引入卷积代替MLP 中的点积运算,近年来在GS 研究中逐渐受到关注。如Ma 等[28]开发了基于CNN 的DeepGS,对2 000 份小麦品种的8 个性状表型进行预测,取得了一定的成效,近期Wang 等[9]更是基于CNN 开发了新的方法DNNGP,并在多组数据集中取得了成功。
集成学习(ensemble learning,EL)[29]通过构建并结合多个机器学习器来完成学习任务,如随机森林(random forest,RF)[30]、GBDT[31]、XGBoost、LightGBM[32]和其他形式的模型融合方法。RF 使用决策树作为弱学习器,在每个决策树的训练过程中,除了采用自助采样法对样本进行采样,还在每个节点的特征选择时随机选取一部分特征进行考虑,其最终的预测结果是基于所有决策树的投票或平均。GBDT 是一种迭代决策树算法,通过使用加法模型,不断减小训练过程产生的残差实现分类或回归。XGBoost 在GBDT 基础上进行了一系列优化,加入了二阶导数信息和正则项等。LightGBM 是微软开发的轻量级梯度提升机,相对XGBoost 具有训练速度快和内存占用低等优点,Yan 等[33]将其用于玉米的一组大型数据集,在预测精度、模型稳定性和计算效率方面展示出了卓越的性能。
相对于线性模型和贝叶斯方法,机器学习模型能够更好地对基因型和表型之间的非线性关系进行学习,不过其缺点是可解释性往往较差,难以对生物样本的遗传效应组成进行分解,以及对各个位点的效应进行评估。如何增强模型对遗传效应的解析能力,提高优异等位基因聚合的效率,是未来机器学习方法研究所面临的一项重要挑战。
2 全基因组选择功效的影响因素
大多数GS 研究使用待测群体表型预测值与实际值之间的相关系数或决定系数衡量模型的预测精度,以反映GS 的功效。作物GS 的实际功效受到多种因素的影响,其中遗传因素包括目标性状遗传力、训练群体和育种群体间的关系、标记密度、标记和QTL 间连锁不平衡的程度等,非遗传因素包括训练样本数量、模型和算法及其参数的选择,以及数据的清洗方案等(表1)。

表1 影响全基因组选择功效的因素及其优化策略Table 1 Factors affecting GS efficacy and corresponding optimizating strategies
2.1 遗传因素
研究表明,预测精度首先受到目标性状遗传力的影响,遗传力越高,精度越高[12]。作物的产量性状容易受到环境等非遗传因素的影响,往往具有较低的遗传力,然而幸运的是,这并不意味着GS 的低效。Wang 等[34]在水稻中的研究结果表明,GS优选群体的平均选择优势与性状的遗传力并无直接联系。虽然产量等性状的预测精度较低,但是其原因在于高占比的误差方差,这并不妨碍育种家利用GS 技术获得理想的遗传增益。对于较低遗传力的性状,适当扩大优选群体,就能够获得稳定的较高平均选择优势。
此外,训练群体和育种群体之间的关系也会影响选择的效果,有研究表明,与训练样本遗传上相似的群体能够获得较高的预测精度,对于一些遗传不相似的亚群,则预测精度较低[35]。在Wang 等[6]对玉米亲本GCA 的预测研究中,参与训练集田间试验的亲本相对未参与者能够获取更高的预测能力,也提示了训练集与测试集之间紧密遗传关系对模型预测的积极贡献。然而大量增加与测试群体遗传相似的训练样本,可能降低优选品种的遗传多样性,从而不利于长期的遗传增益。因此,在实际育种中需要寻求训练集和测试集之间关系的平衡[36]。
GS 假设基因组上总有标记和影响性状的QTL之间存在连锁不平衡,增加标记的密度能增加标记和QTL 之间的LD 程度,从而可能获得更高的准确度[37]。理论上,标记密度越大越好,但是与训练种群的数量相比,其对预测精度的影响较小[38]。在Wang 等[34]使用GBLUP 方法对水稻的GS 预测中,1610K 标记的预测精度高于470K,又高于96K,不过其差异非常微小,即标记密度达到一定程度后,GS 的精度难以显著提高。因为高密度标记的获取成本较高,且给数据的预处理和模型训练带来困难(如GBLUP 方法的G 矩阵运算需要超出一般个人电脑配置的更大内存,选择收缩算法的变量选择难度加大和训练速度的大幅降低),所以在实际的作物GS 应用中,根据我们的经验,使用全基因组上均匀分布的数万个SNP 标记具有较高的性价比。此外,标记和QTL 间的LD 程度也会影响GS 的准确性,随着世代的增加,标记和QTL 的LD 会逐渐降低。Meuwissen 等[1]发现在基因型测定后的前2 个世代GS 的准确性下降较快,其他世代下降速度则相对减慢。随着世代的增加,遗传力较高性状的基因组预测准确性降低较慢。
2.2 非遗传因素
样本数量和GS 模型等非遗传因素也会对预测效果产生影响。较大的训练样本十分有利于GS 模型对等位基因效应的准确估计,进而有利于对潜在品种的精确选择。尤其是对低遗传力的性状,增大样本数量和试验重复数可以降低误差效应的不利影响,提高模型的功效。前人的研究表明,遗传力为0.2的性状需要的训练样本数量超过1 000[39]。
实际的GS 过程中,模型和算法是更易调整的可变因素。不过其挑战在于,虽然有大量的GS 方法可供选择,但是育种家在使用GS 技术时仅能对少数方法的预测结果开展进一步的田间鉴定。研究者在育种组合的优选之前,首先需要对GS 方法进行优选,所以GS 方法的比较研究是十分重要的基础工作。近年来一些学者使用不同的作物群体数据,对多种GS 方法进行了比较。Xu 等[38]利用一组基于NCII 设计的水稻数据集,比较了6 种GS 方法的表现,发现不同方法的可预测性存在显著差异,其中GBLUP 和LASSO 最佳,SVM 和部分最小平方法最差。Wang 等[12]以一组小麦数据集为基础,利用6 种方法进行了模拟研究,并用于小麦实际产量数据的预测。其结果表明,对于具有不同遗传结构的性状,各GS 方法的表现差异明显。基于贝叶斯的选择收缩算法对QTL 的数目较为敏感,当性状由较少数目的QTL 控制时,预测精度较高,当影响数量性状的QTL 数目很多时,精度则会下降。GBLUP 和RR‑BLUP 的稳健性较强,其预测精度不受QTL 数目的影响,在预测作物产量等由大量微效基因决定的性状时,更具优势。近年来一些学者使用新的机器学习模型和算法,在作物的GS 中取得了令人瞩目的成绩[8-9,33],不过各种机器学习方法的功效是否受性状遗传结构的影响,还缺少相关的研究。此外,超参数的选择对一些机器学习方法的性能影响较大,如深度学习中网络层数、神经元数目、滤波器大小、迭代次数和激活函数的不同调优方案,可能产生完全不同的预测效果。因此使用多组不同的数据集,进行网格搜索、随机搜索或人工经验调参以优化参数组合,对于提高模型的精度和泛化能力是十分重要的工作。
尽管一系列模型和算法先后被提出,并使用不同的数据集进行了广泛的比较,但是当前的GS 建模仍然面临“大p,小n”问题,即标记数目远大于样本量,容易导致多重共线性和过度拟合,进而影响模型表现的稳定性,以至于没有哪种方法在大多数情况下都能保持领先的预测精度。Xu 等[40]建议在使用GS 方法辅助育种决策之前,先在训练集中利用交叉验证对比不同模型的精度,以实现GS 方法的优选。不过实际中待测群体和训练群体之间往往存在一定的遗传差异,要从一般意义上解决上述问题,建立科学的数据清洗方案可能是一种有效的途径。除了常规的标准化或归一化等预处理,还应开发与基因组信息特征相适应的降维方案,采用单倍型划分或主成分分析等方法,在保留大部分标记信息的基础上大幅减少自变量数目,降低过拟合的风险,以提高GS 中众多选择收缩方法的健壮性。这一点对于GS 精度的突破性提升,是至关重要的。
3 全基因组选择方法的拓展
3.1 非加性效应模型研究
传统的GS 方法在估计遗传效应时大多只考虑最简单的加性效应(育种值),虽然非加性遗传效应不能直接从亲本传递给子代,但是它们对那些和适应性紧密相关的性状和低遗传率性状是非常重要的[10]。尤其对于作物的杂种育种,杂交种与亲本之间存在明显的基因表达差异[41-42],表现为加性和非加性等差异表达模式。因此,很多学者提出在GS 中有必要考虑非加性效应[43-45]。Xu 等[46]的模拟研究表明,在混合模型中纳入上位性多基因协方差,可以提高QTL 定位的分辨率,并将其用于水稻产量相关性状的遗传效应解析。
在GS 模型中,Xu 等[2]引入显性和上位性等效应,模拟实验表明能够提高预测能力,不过在预测杂交水稻实际表型值时,新的模型未能获得预期的效果,原因可能是模拟中的部分假定与实际情况存在偏差。另外在样本群体较小的情况下,基于全基因组的变异位点和位点间互作进行分析时,超饱和模型难以保证估计的精度。因此,如何对基因型值进行科学编码以正确反映显性和上位性等遗传效应,是非加性模型构建所面临的重要挑战。近期Miranda等[47]的研究就借助Huang 等[48]提出的关系矩阵构建方案,开发了用于GBLUP 模型加性和显性效应参数评估的方法,不过模型的预测效果仍需在更多数据集中研究验证。近期Li 等[49]将GS 中具有加性和/或显性效应的12 个品质性状的遗传基因位点分层,提高了对杂交种预测的准确性,也为非加性效应的估计提供了新的思路。
机器学习方法是实现非加性遗传效应评估的又一重要途径。Budhlakoti 等[50]的研究表明,当模型中加入非加性遗传结构时,SVM 等非参数方法的性能可能比参数方法的性能更好,原因在于这些方法不需要严格的统计假设。王向峰等[51]提出,为了克服传统混合线性模型基因组预测的不足,应用机器学习,尤其是深度学习等人工智能领域中的先进算法,是GS 育种的下一步发展方向。Wang 等[52]则指出,深度学习算法具有强大的非线性建模能力,有助于提高GS 的精度。近期Wang 等[9]分别对多个数据集,首先使用主成分方法降维,然后基于深度神经网络开展预测,模型精度超过了其他多种方法。从原理出发,以深度学习为代表的机器学习方法能够自主学习基因位点的主效应、等位基因之间或者位点间的互作关系,避免了基于某种简化假设模型的基因型数值再编码,从而有机会更好地捕捉位点的非加性效应。
3.2 群体构建方案研究
GS 方法的育种应用离不开作物群体的科学构建。Guo 等[5]利用玉米、小麦和水稻数据集,研究了预测杂交种表型的训练集设计方案。将杂交种的所有亲本自交系视为需要从中选择杂交组合的整体遗传空间,设计并测试了3 种代表性子集选择方法,以建立用于杂交种基因组预测的训练集。其中PAM方法围绕聚类的中心点进行划分,FURS 方法快速地从给定的图中选择一组代表性节点,MaxCD 方法则在连通性和多样性最大化的基础上进行选择。结果表明,有效的基因组预测模型只需要整个训练集大小的2%-13%,揭示了对海量遗传组合高效推断的可能。Chung 等[53]的研究也指出,在杂种育种过程中,单纯对亲本育种值的优选会导致遗传多样性的丧失,为了保持基因组多样性,在亲本选择过程中应避免选择亲缘关系密切的材料。该研究提出了一种平衡育种值和遗传多样性的折中策略,并在两组水稻数据集中得到了验证,该策略与前述Guo 等[5]的MaxCD 方法有着共通之处。
在科学开展遗传交配设计的基础上,GS 模型还可用于GCA 等育种指标的精确预测。王欣等[54]将NCII 水稻数据集的亲本GCA 看作目标性状,进行了5 倍交叉验证和留一法的基因组预测,结果表明其预测是有效的,能够帮助育种家实现对亲本的科学选择。不过将GCA 当作因变量,首先需要获得所有训练集亲本的GCA 值。尽管NCII 设计能够完全满足这一条件,但是由于成本和田间试验条件的限制,很多情况下作物的组配设计是稀疏的。Wang 等[6]进一步使用SPDC 设计,研究了稀疏条件下利用全基因组标记对玉米亲本GCA 的预测情况。结果表明在训练集杂交种组配异常稀疏的情况,也能够实现对亲本GCA 的精确估计。另一方面,在遗传交配设计时,应尽可能让更多的亲本参与训练集的田间试验,以获取较高的预测能力。
3.3 多性状和多环境预测研究
一般的GS 方法关注单一环境下单个性状的研究。然而,对单个性状的预测和选择忽视了关联性状共同的生物学基础以及多性状的协调发展[55]。综合选择指数方法,是动植物多目标育种选择的常用方法,可以被用来同时改良多个性状。GS 的快速发展,为选择指数带来了新的前景。Schulthess 等[56]使用黑麦中的两个性状建立选择指数,并将其看作单一性状用GS 方法进行预测。Leite 等[57]利用选择指数和多变量分析筛选表型优异的大豆基因。Lyra等[58]将玉米杂交种在不同氮胁迫下的性状组合以构建选择指数,然后用GS 方法进行预测,结果表明方法是有效的。Xiao 等[59]在水稻中通过全基因组测序解析育种群体中有利基因分布以及连锁关系,并结合GS 优化品种改良方案实现了品种多性状的协同提升。
对作物的多个性状进行联合分析,还能够提高对目标性状预测的精度[56]。Wang 等[34]基于NCII设计的水稻数据集,利用指示变量构造的关系矩阵反映多变量之间的关系,在性状数据非平衡的情况下(待测群体目标性状之外的部分性状表型已知),两性状联合分析时对性状的预测能力较单性状分析时平均要高6.4%,八性状联合分析时较单性状分析平均要高26.7%。不过在性状数据平衡情况下(待测群体所有性状表型未知)的一些研究中,多性状模型的精度并非总是优于单性状[27]。通过构造选择指数也可以实现多性状的联合预测,针对性状数据平衡的情况,Wang 等[60]提出了一种基于选择指数的多性状GS 方法,该方法利用与目标性状相关的多个辅助性状及其蕴含的目标性状遗传信息构建选择指数,不仅能实现对水稻杂交种多个性状的综合选择,还能对目标性状进行辅助预测,提高了低遗传力目标性状的预测精度。近期Liang 等[61]提出了一个机器学习框架MAK,通过构建多目标集成回归链和自动选择辅助性状来提高目标性状的预测精度,该框架仅使用待测样本的基因型信息预测目标性状育种值。在4 个真实的动植物数据集中,其预测能力显著高于GBLUP 和多种贝叶斯方法。
植物表型是由基因型、环境型和基因型与环境相互作用的综合作用决定的[48]。作物育种中大量表型数据的观测值来自多年多点的不同环境,育种家希望预测的不仅是潜在材料的育种值,还包括特定环境下的表型值。Lopez‑Cruz 等[62]将G × E效应纳入GBLUP 模型,显著提高了模型的预测能力。Cuevas 等[63]进一步将非线性高斯核与Lopez‑Cruz 等的基因环境互作模型相结合,发现模型对CIMMYT 小麦数据集的预测能力提高了17%。贝叶斯模型也同样被扩展为基因环境互作模型,在小麦和玉米中取得了高于单环境的预测精度[64-65]。近期Rogers 等[66]在玉米中的研究表明,使用环境协变量的基因组预测能力取决于训练集和测试集数据之间环境的相似性。相较于遗传相似性,数据集之间的环境相似性对预测效果影响更大。Yan 等[67]则指出,如果确定了可重复的基因环境互作模式,则必须将作物目标区域划分为子区域或大环境。育种和大环境特异性品种的利用会将可重复的基因环境互作转化为大环境内的基因型主效应,从而提高选择的增益和可靠性。如果没有发现可重复的基因环境互作模式,则必须将目标区域视为单个大环境,通过充分测试来适应基因环境互作。上述多项研究结果提示,在进行多环境的联合GS 过程中,首先明确大环境的划分,继而将同一大环境内尽可能多的表型观测信息纳入模型,是一种行之有效的策略。
3.4 多组学预测研究
一般的GS 方法忽略基因组与其下游调节因子之间的相互作用[68]。下游的转录组、蛋白组和代谢组等组学信息是由基因型向表型传递的中间产物,它们反映了不同生物层内部和之间的相互作用[69]。随着组学技术的进步,代谢组学和转录组学数据为作物的表型预测提供了新的来源。一些研究使用亲本转录组或代谢组学数据预测待测杂交种的表现。Frisch 等[70]首次使用21 个亲本自交系的表达谱数据和98 个杂交种的表型数据对玉米杂交种进行了预测。基于相同的数据集,Fu 等[71]使用56K 微阵列分析亲本自交系的基因表达,发现杂交种的表现可以通过亲本自交系的基因表达数据得到准确预测。Zenke‑Philippi 等[72]使用2K 的核心基因表达数据和1K 的AFLP 标记数据对玉米杂交种的产量和干物质含量进行转录组和基因组预测。在使用岭回归模型时,对杂交种表型的转录组预测略好于基因组预测。对于代谢组学预测,Riedelsheimer 等[73]利用285 份玉米自交系的56 110 个SNP 和130 种代谢产物,以及570 份测交种的表型数据构建GS 模型,预测了7个性状的一般配合力,发现代谢物的预测精度与基因标记的预测精度相当。Xu 等[74]利用210 份水稻亲本的代谢组数据预测278 份杂交种的产量,发现与基因组预测相比,预测能力几乎提高了一倍。
多组学数据的联合预测有可能进一步提升预测的效果。Guo 等[75]使用玉米数据评估了基因表达和代谢数据在基因组预测中的效果,其研究结果表明,基于基因表达和代谢产物的预测能力是特异性的,受到测量时间、组织样本以及基因和代谢产物数量的影响。不过与仅使用全基因组标记的GBLUP 模型相比,将基因表达水平和代谢物丰度与遗传标记相结合显著提高了预测能力,有助于提高复杂性状的遗传增益。Westhues 等[69]将玉米转录组数据与亲本自交系的基因组数据相结合,发现能够提高对潜在杂交组合预测的成功率。Schrag[76]等也利用玉米亲本系的基因组、转录组和代谢组数据,评估了基于这些组学数据对待测杂交种的预测能力,发现预测因子和性状的预测能力之间存在很强的互作关系,信使RNA 是产量和干物质含量的最佳预测因子,结合信使RNA 和基因组数据作为预测因子,在两个性状上都有很高的预测能力,提示下游的组学数据是基因组预测的重要补充,有助于对潜在杂交种的精确选择。Wang 等[77]对水稻不同组学数据组合后的预测能力进行了比较,得出的结论是,使用基因组和代谢组学数据组合的预测通常比单一组学预测或基于其他组学数据组合的预测效果更好。Wu 等[78]在大麦中也发现,来自转录组和代谢组的任何预测因子在3 个性状上的平均预测能力都高于SNP 标记,并建议使用集成的组学数据集开展预测工作。
转录组和代谢组相较基因组更接近生物的表型,其数据的充分使用有利于预测精度的提高,不过将其用于育种实践的困难是,数据获取成本相对高昂,且杂交种的转录组和代谢组都难以像基因组一样直接从亲本的组学信息中精确推断,其预测能力可能显示出对性状的特异性。相对于组学数据,单交种双亲的表型信息更容易在早期以较低的成本获取。近期Xu 等[7]提出了将作物亲本表型信息纳入杂交种表型预测的策略,为基于多元数据的预测提供了新的途径。该研究基于210 份水稻自交系的基因组、转录组和代谢组数据以及278 份杂交种的表型数据,利用混合线性模型,进行了多组学的联合分析,并整合亲本表型预测杂交种的表现。研究结果表明,无论采用何种组学信息进行预测,结合双亲信息后,所有性状的预测准确性均有不同程度提高,产量、穗粒数、分蘖数和千粒重的平均预测力分别提高了13.6%、54.5%、19.9%和8.3%。
4 全基因组选择育种芯片研发现状
近年来,作物SNP 育种芯片的不断研发,为GS 中基因型数据的获取提供了重要技术条件。目前超过25 种作物中已经开发了百余款芯片[79-80],其中水稻的代表性芯片有RICE6K 和RiceSNP50 等;玉米代表性芯片有MaizeSNP600K、MaizeSNP50 Beadchip 和Maize6H‑60K 等;小 麦 代 表 性 芯 片 有Wheat 9K iSelect、Wheat 90K iSelect、Wheat 660K Axiom 和Wheat HD Genotyping Array 等;马铃薯代表性芯片有SolSTW array 等;大豆代表性芯片有SoySNP50K 和SoyaSNP 180K Axiom 等。这 些 芯 片主要是基于国外的Illumina Infinium BeadChip 技术或Affymetrix Axiom 技术。我国科学家建立了具有自主知识产权的靶向测序-液相芯片技术,并在水稻、玉米和小麦上分别开发了GenoBaits® Rice 40K、GenoBaits® Maize 45K 和GenoBaits® WheatSNP16K 等一系列液相芯片。
上述芯片虽然在种质资源遗传多样性评估、品种指纹图谱构建和重要基因的定位中具有重要用途,但是要针对实际育种群体高效开展GS 育种还存在诸多困难:(1)现有芯片信息覆盖度不高,不利于持续提高GS 的效率。目前的GS 研究大多都是基于SNP 标记,忽略了很多与性状关联但与邻近SNP 无连锁的结构变异,而这些结构变异与抗逆性、抗病性、产量和品质等重要性状有关,其鉴定工作对于作物育种有着重要的意义,但是目前作物芯片中尚未包含这类结构变异信息,从而造成遗传力的丢失。(2)现有芯片通用性不足,不同基因型数据难以共享。GS 的准确性随着训练群体的增大而增加,然而即使是同一作物,不同的育种家往往也会针对各自的群体和育种目标选择不同的育种芯片,造成群体间不同位置的标记无法纳入同一预测模型,这极大地阻碍了作物基因型数据的共享,限制了GS 预测模型的优化和准确性的提高。(3)对于大规模育种应用,芯片检测成本仍然较高。作物育种群体数量庞大,开展GS 育种时需要考虑基因型鉴定成本。尽管目前SNP 芯片的成本已经有所降低,但是单个样本分析的成本仍需百元左右,且标记密度不同会导致较大的成本差异,无法满足现代作物育种的低成本需求,大多数育种企业囿于巨额的基因型鉴定成本投入而无法大规模应用GS 育种技术。(4)缺乏育种芯片专用的分析软件和平台。TASSEL 和PLINK 等主流基因型分析软件只能支持特定格式的输入文件,如Hapmap 或VCF 格式。育种家手中的芯片数据往往具有各种不同的格式,目前尚缺乏此类芯片数据的标准化分析工具。此外,多数种企和育种单位的信息化水平仍然较低,缺乏系统的育种芯片处理及育种决策软件和平台。(5)我国底盘技术创新不足,核心技术受制于人。目前市场上的SNP 芯片主要以Illumina 公司和Affymetrix 公司的技术为主,我国缺乏底盘技术的自主知识产权,随时面临技术“卡脖子”的风险。
要克服上述困难,只有充分利用功能基因组学研究成果,研发具有我国自主知识产权、广适性好的作物育种专用芯片。在考虑已克隆的高产、优质、抗病虫、抗逆、养分高效等重要性状功能基因和关联SNP 标记的基础上,整合相关结构变异标记,提高育种芯片的检测功效。并开发与育种专用芯片配套的数据分析软件,以图形化界面的方式完成对种质资源类型的划分、全基因组选择模型的构建、预测模型的优化、预测准确性的评估,对测试群体表型进行快速、精准预测,实现对作物产量、品质、抗性等重要位点的快速筛查。
更进一步,应当构建智能决策育种平台,提升育种效率和决策水平。作物广泛来源的(包括地方品种、亚种和品系等)丰富遗传变异,可以通过基于基因组信息的人工智能和大数据等现代技术来识别和发现[36]。具体措施,应广泛收集表型、基因型和环境数据,同时制定数据管理的标准与规范,强化遗传育种与人工智能和大数据等信息技术的交叉集成,协同建立通用的智能决策育种平台,通过平台、技术、群体、数据、模型以及育种材料的充分共享和积累,实现资源利用和育种效率的最大化,创新发展以育种专用芯片应用为核心技术的GS 育种体系,为作物育种的精准化、高效化、智能化发展提供有力支撑。
5 全基因组选择育种展望
GS 育种技术的逐渐成熟和广泛应用为作物育种研究提供了新的机遇,将其与重要目标性状基因的精准鉴定结果相结合,有望大大加快优异基因聚合的效率,并创制出更加丰富的遗传资源。尤其针对我国作物育种群体遗传来源较为狭窄的问题,利用基因组水平上的精准预测,能够帮助育种家放眼更广泛来源的种质材料,通过精确预测和育种方案的科学设计,聚合更多的有利等位基因,以创建作物的优异育种新材料。如果将基因组、转录组和代谢组等组学信息相结合,配合对作物多个性状的联合预测,有望实现作物多个性状之间的协调发展,为培育适应机械化生产、优质高产多抗广适作物新品种提供有效途径。
虽然GS 技术在作物育种中的应用前景广阔,但是其发展仍然面临着众多挑战,主要包括以下几点:(1)一般的GS 方法只考虑加性效应,部分学者将显性及上位性等效应纳入模型,但是预测效果还不够理想;(2)前人的GS 研究大多只针对特定环境下特定作物群体的单个性状,忽视了关联性状共同的生物学基础以及多性状的协调发展,且缺乏详细的环境组学数据,难以实现对基因环境互作模式的识别与利用;(3)多数GS 研究只用到基因组信息,多组学信息和研究成果没有得到充分利用;(4)缺少GS 育种专用芯片以及配套软件和平台,数据共享程度低,限制了GS 效率的提高;(5)作物领域中已有的GS 研究很多停留在方法探索阶段,未能广泛付诸于实际的育种工作。
针对上述问题,首先应结合已有的生物学和遗传学研究成果,遴选作物全基因组上目标性状的候选基因,开发与基因组信息特征相适应的降维方案,以大幅降低模型中的变量数目,同时应用人工智能领域中的先进算法,提高对各类非加性遗传效应的准确预测;第二,广泛收集表型、基因型和环境数据,并对模型进行优化,注重作物多个性状之间的协调发展,识别并利用基因环境互作模式,提高选择的增益和可靠性;第三,应结合人工神经网络,机器学习等最新的数学方法,积极开展作物多组学预测研究,构建多组学信息与目标性状之间的数量遗传模型,提高多组学联合预测的效果;第四,可以谋划构建GS 专用芯片和统一的GS 平台,实现群体之间的信息共享与利用,提高数据的利用率;第五,GS 研究必须结合农业发展的实际情况与切实需求,让理论和方法研究更好地服务于实际育种工作,为培育适应机械化生产、优质高产多抗广适作物新品种提供高效途径。总之,随着作物育种精准化和智能化的需求不断提升,以及基因组学和人工智能技术的快速发展,未来的GS 研究工作充满了机遇和挑战。
猜你喜欢
区域治理(2022年40期)2022-11-27
今日农业(2021年11期)2021-08-13
动漫界·幼教365(小班)(2019年10期)2019-10-28
动漫界·幼教365(大班)(2019年10期)2019-10-28
动漫界·幼教365(中班)(2019年10期)2019-10-28
现代园艺(2017年21期)2018-01-03
中国康复理论与实践(2015年10期)2015-12-24
医学研究杂志(2015年5期)2015-06-10
现代检验医学杂志(2015年5期)2015-02-06
遗传(2014年3期)2014-02-28
