
WIN55212-2通过调控mTOR/HIF-1α/PFKFB3信号通路抑制糖酵解并减轻脓毒症小鼠急性肺损伤*
2024-04-07 12:11段倩雯董旭鹏马玉清
中国病理生理杂志 2024年3期
段倩雯, 董旭鹏, 马 源, 刘 澈, 张 铭, 马玉清
(1兰州大学第一临床医学院,甘肃 兰州 730000;2兰州大学第一医院麻醉科,甘肃 兰州 730000)
脓毒症(sepsis)是一种由宿主反应失调导致的危及生命的器官功能障碍[1]。在脓毒症发展过程中,急性肺损伤(acute lung injury, ALI)/急性呼吸窘迫综合征(acute respiratory distress syndrome, ARDS)是最常见的器官损伤,ALI/ARDS 患者预后较差,死亡率高达35%~45%[2],目前仍然是一个医疗危机。
巨噬细胞活化是脓毒症的主要特征,巨噬细胞活化通过Warburg效应(也称为有氧糖酵解或代谢重编程)诱导过度增殖和炎症反应[3]。6-磷酸果糖-2-激酶/果糖-2,6-双磷酸酶3(6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3, PFKFB3)是PFKFB 家族的同工酶,被称为糖酵解调节剂,是糖酵解关键酶磷酸果糖激酶1 的有效变构调节因子,可维持高糖酵解速率[4]。
哺乳动物雷帕霉素靶蛋白(mammalian target of rapamycin, mTOR)作为一种非典型丝氨酸/苏氨酸蛋白激酶,是巨噬细胞活化的重要位点[5]。mTOR 激活促进缺氧诱导因子1α(hypoxia-inducible factor-1α,HIF-1α)的转录及翻译,调控促炎因子并驱动多种糖酵解酶的表达,促进糖代谢从氧化磷酸化转变为糖酵解,为细胞增殖提供能量[6]。研究证实,在巨噬细胞中敲除mTOR相关基因,转录因子HIF-1α 表达降低,从而减少葡萄糖摄取,抑制糖酵解[7]。
WIN55212-2(WIN)是一种非选择性具有抗炎特性的大麻素受体1 和大麻素受体2 激动剂。研究表明,WIN 可以激活大麻素受体2,减少巨噬细胞TNFα 和活性氧的生成产生抗炎和抗氧化作用[8]。Feng等[9]证实,WIN可以通过调节p38MAPK信号通路,降低结肠炎小鼠血浆TNF-α、白细胞介素6(interleukin-6, IL-6)及肠组织MPO 活性,并上调claudin-1 表达,改善肠道屏障功能,减轻肠道炎症反应。WIN 可否通过抑制糖酵解,减轻脓毒症ALI 及其与mTOR/HIF-1α/PFKFB3 信号通路之间的关系,尚未有研究报道。因此,本研究拟建立脂多糖(lipopolysaccha‐ride, LPS)诱导的小鼠脓毒症模型,探讨WIN 是否通过调控mTOR/HIF-1α/PFKFB3 信号通路,抑制糖酵解,缓解脓毒症小鼠ALI。
材料和方法
1 动物
6~8 周龄SP F 级雄性C57BL/6J 小鼠24 只,体重18~22 g,购自兰州大学医学院动物实验中心,动物许可证号:SCXK(甘)2018-0002。遵循实验动物3R 原则。小鼠正常进食,自由进水,人工光照和黑暗时间12 h 交替。本实验符合相关动物伦理法规,获得兰州大学第一医院动物伦理委员会批准,审批号:LDYYLL2023-319。
2 主要试剂与仪器
LPS(Sigma); WIN55212-2(Cayman);mTOR 激动剂MHY1485(MCE);磷酸盐缓冲液(PBS)、4%多聚甲醛固定液(Wabcan);乳酸、乳酸脱氢酶A(lac‐tate dehydrogenase A, LDHA)、IL-1β 和IL-10 ELISA试剂盒(上海酶联生物科技有限公司);磷酸化mTOR(p-mTOR)抗体和β-actin 抗体(北京博奥森生物科技有限公司);HIF-1α 抗体和PFKFB3 抗体(Abcam);RIPA 蛋白裂解液(北京索莱宝公司);二甲基亚砜(DMSO)、蛋白Marker和ECL发光液(Biosharp)。
光学显微镜(Olympus);离心机(Beckman);酶标仪(Thermo)。
3 实验方法
3.1 动物分组及造模 将24 只小鼠随机分为对照(control)组、LPS 组、LPS+WIN 组 和LPS+WIN+MHY1485 组,每组6 只。造模前禁食12 h,自由进水,腹腔注射LPS(10 mg/kg[10])制备脓毒症小鼠模型,LPS+WIN 组造模前30 min 腹腔注射1 mg/kg WIN[11],LPS+WIN+MHY1485 组造模前1 d 腹腔注射10 mg/kg MHY1485[12],并在造模前30 min 腹腔注射1 mg/kg WIN 和10 mg/kg MHY1485,control 组和LPS组在上述相同时点腹腔注射等体积DMSO 及生理盐水。造模24 h 后观察小鼠毛发、活动和精神等一般状况,有无竖毛及眼角分泌物;摄食、进水及攀爬打斗等行为的活跃程度;对声音、触碰等刺激的行为反应;呼吸频率等判断造模是否成功。水合氯醛4 mL/kg腹腔注射麻醉小鼠后,之后将其仰卧于固定板上;剪去心前区毛发并消毒,以食指指腹定位小鼠心脏搏动区;手持1 mL 注射器穿刺心搏最强处,收集足量血液标本。静置30 min 后以3 000 r/min 离心15 min,取上清液于−80 ℃冰箱储存。心脏取血后处死小鼠。常规剪去毛发、消毒,暴露胸腔;取肺组织,PBS冲洗3遍,取右肺下叶固定于4%多聚甲醛溶液,用于病理学观察;分装剩余标本,−80 ℃冰箱储存,用于后续检测。
3.2 肺指数检测 取肺脏吸干表面水分后称重,计算肺指数[13]。肺指数=肺湿质量(mg)/小鼠体质量(g)×100%。
3.3 观察肺组织病理结构 取肺组织,置于4%的多聚甲醛中固定48 h,脱水后进行石蜡包埋、切片、染色、显微镜下观察肺组织结构变化,肺组织Smith评分[14]按照肺水肿、炎症细胞浸润、肺不张、出血、坏死和透明膜形成进行0~4分的半定量分析。
3.4 ELISA 法检测肺组织炎症因子IL-1β、IL-10 及血清乳酸、LDHA 表达水平 肺组织称重量,按照1 g加入5 mL PBS 的比例加入PBS,在冰水浴中尽量剪碎组织,随后按照每20 mg 肺组织加入150~200 µL RIPA 裂解液的比例加入裂解液,在冰上使用匀浆机制备成10%肺组织匀浆。将制备好的组织匀浆于4 ℃、3 000 r/min离心10 min,离心后的匀浆取上清液进行分装,−80 ℃放置保存。按照ELISA 试剂盒说明检测肺匀浆IL-1β 和IL-10 表达水平。取血清上清液,按照ELISA 试剂盒说明检测血清乳酸和LDHA水平。
3.5 Western blot 检测mTOR/HIF-1α/PFKFB3 信号蛋白的表达水平 取−80 ℃肺组织匀浆,加入RIPA组织裂解液1 mL,裂解30 min,在4 ℃、12 000 r/min离心10 min,转移上清至新的离心管提取组织蛋白。加热变性10 min,经十二烷基硫酸钠-聚丙烯酰胺凝胶电泳100 min,转移至聚偏二氟乙烯膜上,洗膜后加入Ⅰ抗(p-mTOR、HIF-1α、PFKFB3和β-actin抗体,1∶2 000 稀释),4 ℃孵育过夜。次日洗膜,加入辣根过氧化物酶标记的山羊抗兔Ⅱ抗(1∶100 000 稀释)37 ℃摇床孵育2 h,TBST 充分洗膜。增强化学发光法显色曝光,采集图像后用ImageJ 软件分析各组条带灰度值,用目标蛋白与内参照蛋白的灰度值比值反映目标蛋白表达量。
4 统计学处理
所有数据采用Graphpad Prism 9.5 软件进行统计学分析并作图,所有数据均以均数±标准差(mean±SD)表示,本研究实验数据均为计量资料且符合正态分布,多组间比较采用单因素方差分析,两组间比较采用LSD-t检验。以P<0.05为差异有统计学意义。
结 果
1 小鼠一般状态
LPS造模24 h后,control组小鼠精神状态、活动、饮食、呼吸及对刺激的反应较造模前无明显改变,LPS 组小鼠精神萎靡,进食进水减少、寒战、呼吸急促,眼角分泌物增多,竖毛,LPS+WIN 组与LPS+WIN+MHY1485 组较LPS 组小鼠上述表现轻微,而LPS+WIN+MHY1485组相较于LPS+WIN 组小鼠上述表现加重。
2 各组小鼠肺指数比较
Control 组、LPS 组、LPS+WIN 组 及LPS+WIN+MHY1485 组小鼠体质量比较差异均无统计学意义。与control 组相比,LPS 组湿肺质量及肺指数增加(P<0.05);与LPS 组相比,LPS+WIN 组相较于LPS 组湿肺质量及肺指数明显降低(P<0.05);LPS+WIN+MHY1485 组相比于LPS+WIN 组湿肺质量及肺指数增加(P<0.05),见表1。
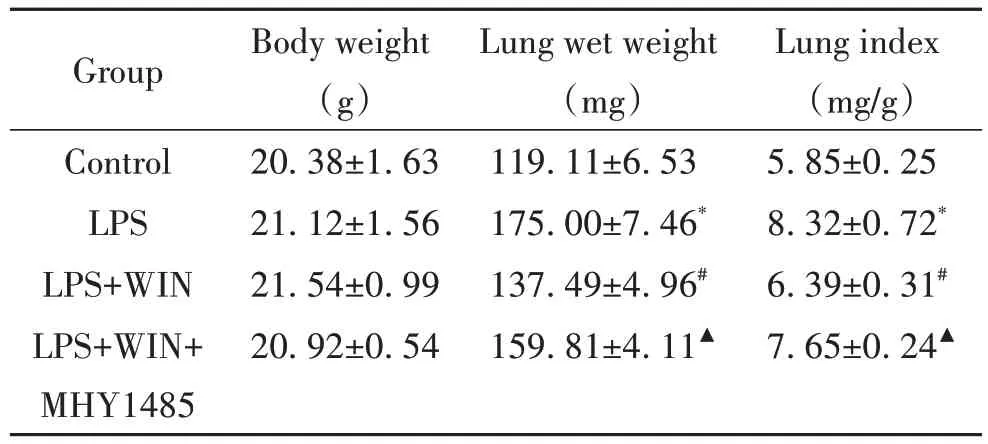
表1 各组小鼠肺指数比较Table 1. Comparative analysis of lung index in mice of different groups (Mean±SD. n=6)
3 各组小鼠肺组织病理结构改变
光镜下,control 组肺组织结构完整,肺泡及间质未见水肿及明显炎症浸润,LPS 组肺间质水肿,肺泡明显萎陷,坏死组织出现,肺泡和间质可见大量炎症细胞浸润,肺泡腔可见出血及透明膜形成;LPS+WIN组肺泡充血、出血、炎症渗出、肺泡壁增厚和透明膜形成等损伤有所减轻;LPS+WIN+MHY1485 组肺水肿和肺不张较LPS+WIN 组加重,肺泡及间质炎症细胞浸润增多,肺泡出血增加。与control 组相比,LPS组Smith 评分显著增加(P<0.05);与LPS 组相比,LPS+WIN 组Smith 评分显著降低(P<0.05) ;LPS+WIN+MHY1485 组较LPS+WIN 组相比Smith 评分增加(P<0.05),见图1。
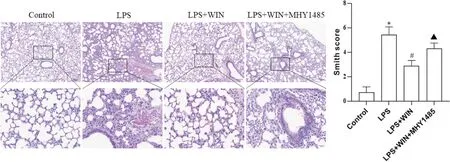
Figure 1. Pathological alterations and Smith score of lung tissues in mice of different groups( HE staining, scale bar=100 or 20 µm).Mean±SD. n=3. *P<0.05 vs control group; #P<0.05 vs LPS group; ▲P<0.05 vs LPS+WIN group.图1 各组小鼠肺组织病理学变化及Smith评分
4 各组小鼠肺组织IL-1β和IL-10水平
ELISA 检测各组小鼠肺组织匀浆炎症因子IL-1β和IL-10的表达情况,结果显示:与control组相比,LPS 组中IL-1β 表达升高,IL-10 表达下降(P<0.05);与LPS 组相比,LPS+WIN 组在WIN 预处理后IL-1β表达相对下降而IL-10 表达升高,差异均有统计学意义(P<0.05)。此外,LPS+WIN+MHY1485 组与LPS+WIN 组相比,IL-1β 表达升高,而IL-10 表达下降(P<0.05),见图2。
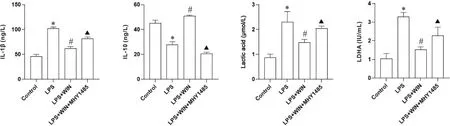
Figure 2. Levels of IL-1β and IL-10 in lung tissues, and lactic acid and LDHA in the serum of mice in each group. Mean±SD. n=3.*P<0.05 vs control group; #P<0.05 vs LPS group; ▲P<0.05 vs LPS+WIN group.图2 各组小鼠肺组织IL-1β、IL-10及血清乳酸、LDHA水平
5 各组小鼠血清乳酸和LDHA水平
ELISA 检测各组小鼠血清中糖酵解关键指标表达,结果显示:与control 组相比,LPS 组中乳酸、LDHA 表达显著增加(P<0.05);而LPS+WIN 组与LPS 组相比,乳酸、LDHA 表达均下降(P<0.05)。LPS+WIN+MHY1485 组 与LPS+WIN 组相比,乳 酸、LDHA表达增加(P<0.05),见图2。
6 各组小鼠肺组织mTOR/HIF-1α/PFKFB3 信号通路蛋白变化
Western blot检测各组小鼠肺组织p-mTOR、HIF-1α、PFKFB3 表达水平,结果显示:与control 组相比,LPS 组小鼠肺组织p-mTOR、HIF-1α、PFKFB3 蛋白表达显著增加(P<0.05); LPS+WIN 组与LPS 组相比,p-mTOR、HIF-1α、PFKFB3 蛋白表达均下降(P<0.05);LPS+WIN+MHY1485 组与LPS+WIN 组相比,p-mTOR、HIF-1α、PFKFB3 蛋白表达增加(P<0.05)。见图3。
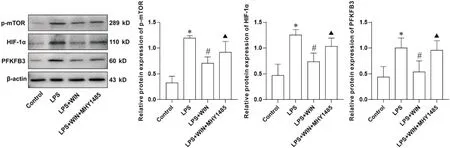
Figure 3. The protein levels of p-mTOR, HIF-1α and PFKFB3 in the lung tissues of mice in each group. Mean±SD. n=3. *P<0.05 vs control group; #P<0.05 vs LPS group; ▲P<0.05 vs LPS+WIN group.图3 各组小鼠肺组织p-mTOR、HIF-1α和PFKFB3蛋白水平
讨 论
在本研究中,我们采用腹腔注射LPS 建立小鼠脓毒症ALI 模型,造模24 h 后,LPS 组小鼠进食进水量减少,出现精神萎靡、寒战、呼吸急促、眼角分泌物增多、竖毛等症状,肺组织病理学变化出现间质水肿,肺泡明显萎陷,坏死组织出现,肺泡和间质可见大量炎症细胞浸润,肺泡腔可见出血及透明膜形成,肺指数增加,表明小鼠脓毒症ALI模型制备成功。
在脓毒症ALI 发展过程中,炎症反应失调是脓毒症ALI 的主要特征[15]。WIN 是一种具有抗炎特性的大麻素受体激动剂,本研究发现,LPS+WIN组肺组织病理学结果显示炎症浸润、出血及肺泡塌陷等损伤减轻,肺指数降低,表明WIN 对脓毒症ALI 具有一定保护作用,激活大麻素受体2 可以缓解脓毒症ALI[16]。也有研究报道,WIN可逆转LPS刺激后TNFα、IL-1β 和IL-6的水平,抑制巨噬细胞的M1型活化,发挥抗炎作用[17],这些研究与本研究结果相一致。
LDH 是糖酵解及糖异生的重要酶系之一,促进糖酵解可以上调Warburg 效应相关酶LDHA 及乳酸的表达,因此,LDHA 及乳酸被认为是糖酵解的关键生物标志物[18]。本研究观察到,LPS 组乳酸和LDHA表达增加,肺水肿及肺组织病理损伤严重,表明脓毒症ALI 伴随糖酵解水平上升。WIN 处理后糖酵解相关标志物水平下降,同时肺损伤减轻。推测WIN 减轻脓毒症ALI 的机理,可能是通过调控糖酵解,减轻炎症反应。Endo 等[19]研究发现,抑制mTOR/HIF-1α信号轴可以降低CD4+T 细胞中TNF-α、IFN-γ 的产生和LDH的表达。Kong等[20]用LPS刺激小胶质细胞后发现,单羧酸转运蛋白1(monocarboxylate transporter 1, MCT1)可通过HIF-1α/PFKFB3信号轴诱导炎症因子IL-1β、IL-6 表达增加,敲减MCT1可抑制小胶质细胞极化和糖酵解速率,表明MCT1可能通过促进糖酵解来加速LPS 诱导的小胶质细胞M1 极化,产生促炎作用。本研究发现,LPS+WIN+MHY1485 组经过mTOR 激动剂MHY1485 预处理后,脓毒症ALI 小鼠肺组织p-mTOR、HIF-1α 及PFKFB3 表达上升,而LPS+WIN 组上述蛋白的表达下调,推测WIN 可能通过mTOR/HIF-1α/PFKFB3信号通路抑制糖酵解。
糖酵解增加与脓毒症过度炎症反应密切相关,因此,糖酵解的调节成为脓毒症干预的潜在策略[21]。Zhong 等[22]发现,在ALI 期间糖酵解增强,用2-脱氧-D-葡萄糖(2-DG,糖酵解抑制剂)抑制糖酵解后,IL-1β及TNF-α表达降低,说明糖酵解增强后,炎症因子表达升高,损伤加重。IL-1β 的产生和释放依赖于炎症小体的活化。研究发现,IL-1β 可以增加糖酵解速率和PFKFB3 表达,证明PFKFB3 与IL-1β 的产生呈正相关[23]。IL-10 是重要的抗炎因子,通过抑制炎症反应在脓毒症过程中发挥重要作用。本研究显示,LPS+WIN组IL-1β表达下调,抗炎因子IL-10升高,肺组织损伤减轻,而LPS+WIN+MHY1485 组IL-1β 表达增加,而IL-10 水平下降,肺组织损伤加重,呈现出与LPS+WIN 组相反的趋势,表明MHY1485逆转了WIN抑制糖酵解和炎症的作用。因此,推测WIN 可能通过抑制mTOR/HIF-1α/PFKFB3 信号通路,调控糖酵解速率,减少炎症因子的表达,缓解脓毒症ALI。
本实验目前仍存在几个局限性。首先,本实验仅在动物实验水平进行了探讨,未在细胞水平进行验证。其次,WIN 也可能通过其他信号通路发挥作用,仍需进一步深入研究。
综上所述,在LPS 诱导的ALI 模型中,WIN 通过调控mTOR/HIF-1α/PFKFB3 信号通路,抑制糖酵解,降低炎症因子的表达,减轻脓毒症ALI。
猜你喜欢
中南医学科学杂志(2022年6期)2022-12-04
江西中医药(2021年8期)2021-08-10
世界科学技术-中医药现代化(2021年12期)2021-04-19
世界科学技术-中医药现代化(2021年12期)2021-04-19
中国循证心血管医学杂志(2020年11期)2020-01-08
植物研究(2018年4期)2018-07-24
中华老年口腔医学杂志(2016年4期)2017-01-15
中国医学装备(2016年6期)2016-12-01
癌变·畸变·突变(2016年5期)2016-08-22
中医研究(2014年11期)2014-03-11
