
Remote stereocontrol in the (4+2) cycloadditions of 1,7-zwitterions:Asymmetric synthesis of multifunctionalized tetrahydroquinoline derivatives
2024-04-06 06:20ChenChenJinZhouJingJiangYangLiTingMaoChengPengGuZhanWeiHuang
Chinese Chemical Letters 2024年1期
Chen Chen,Jin Zhou,Jing Jiang,Yang Li,Ting Mao,Cheng Peng,Gu Zhan ,Wei Huang
State Key Laboratory of Southwestern Chinese Medicine Resources, School of Pharmacy,Chengdu University of Traditional Chinese Medicine,Chengdu 611137,China
Keywords: Asymmetric synthesis Tetrahydroquinoline Remote stereocontrol Pyrazolone Lewis base catalysis 1,7-Zwitterion
ABSTRACT The scope of stereochemistry recognition usually occurs near the chiral scaffold of a ligand or catalyst.Remote stereocontrol,which can surpass the limits of stereorecognition of remote prochiral centers,has long been a challenging object of great interest in asymmetric catalysis.The current work realized the remote stereocontrol of 1,7-zwitterion intermediates formed from Huang’s o-amino aryl MBH carbonates.With simple and easily accessible β-ICD as the bifunctional catalyst,multifunctionalized tetrahydroquinoline derivatives could be synthesized via (4+2) cycloadditions with excellent enantioselectivity and diastereoselectivity under mild conditions.The strategy possesses broad substrate scope,and three types of electron-deficient enones are successfully applied.Mechanistic studies disclosed the Lewis base-catalyzed reaction pathway,and H-bonding between the catalyst and enones is crucial for long-range stereocontrol.Scale-up reaction and transformations of the tetrahydroquinoline products demonstrated the potential of this strategy.
As one of the most important simple nitrogen heterocycles,tetrahydroquinoline widely exists in numerous natural products and bioactive compounds,including pharmaceuticals [1–5].The bioactive tetrahydroquinoline derivatives usually possess one or more stereocenters in their structures.Therefore,the past decade has seen the rapid development of synthetic strategies for constructing chiral tetrahydroquinolines [6–9].For example,great progress has been made through asymmetric hydrogenations [10–17],Povarov reactions [18–22],(4+2) cycloadditions of 1,4-π-allyl metal complex [23–29],and aza-Michael/Michael cascade reactions[30–35].Despite these advances,developing new strategies for the synthesis of novel tetrahydroquinolines in a highly efficient and stereoselective manner is still highly desired.
Recent decades have witnessed the development of the field of asymmetric catalysis,which has been involved in several Nobel Prizes.Researchers can often use chiral ligands,catalysts,and enzymes to achieve the stereoselective synthesis of target molecules.Nevertheless,the scope of stereochemistry recognition usually occurs near the chiral scaffold of a ligand or catalyst.Remote stereocontrol,which can surpass the limits of stereorecognition of remote prochiral (or chiral) centers,has long been a challenging object of great interest in asymmetric catalysis [36–40].So far,only limited studies on controlling the stereoselectivity of the reaction site more than six bonds away from the chiral catalyst [41–44].
The Morita-Baylis-Hillman (MBH) carbonates have emerged as powerful synthons in constructing diverse chiral cyclic compounds[45–49].In the typical activation model,under the catalysis of chiral Lewis base,the allylic ylides generate and participate in the cycloaddition reactions to afford (3+n) or (1+n) products(Scheme 1a) [50–61].The stereochemistry of reactions initiated at the C1-or C3-position can be induced by the adjacent chiral backbone of the Lewis base [62–70].Recently,the Huang group developed a novelo-amino aryl MBH carbonates,which introduces a nucleophilic N atom at the remote site of the electrophilic site.This synthon could serve as useful 1,4-dipole or 1,6-dipole,and unconventional (6+2) and (4+2) cycloadditions with isocyanate have been achieved by Huang and Li [71–75],providing benzodiazocine and 3,4-dihydroquinazolinone products (Scheme 1b).When a Lewis base catalyst was utilized in the reaction of these MBH carbonates,deprotonation of the amino group would form a 1,7-zwitterion intermediate.Thus,the remote stereocontrol in the cycloaddition reaction of this 1,7-zwitterion can be expected to be a highly challenging task (Scheme 1c).So far,the construction of a remote stereocenter distant from the chiral catalyst has yet to be attained.

Scheme 1.Cycloaddition reactions of MBH carbonates and challenges in remote stereocontrol of 1,7-zwitterions.
In this work,we propose a new methodology for the asymmetric cycloaddition of the 1,7-zwitterion (Scheme 1d).Exploration of chiral bifunctional Lewis base catalysts demonstrates that the secondary interaction of H-bonding is effective in transmitting stereochemical information [76–81].Furthermore,excellent diastereoselectivity and enantioselectivity were achieved in the (4+2) cycloadditions with electron-deficient alkenes,affording chiral multifunctionalized tetrahydroquinoline derivatives [82–87].
We initiated the study by investigating the model reaction of MBH carbonate1awithα-phenylidene pyrazolone2a.The feasibility of the (4+2) cycloaddition reaction was studied by using different racemic Lewis base catalysts,including triphenylphosphine,4-dimethyl aminopyridine (DMAP),and 1,4-diazabicyclo[2.2.2]octane(DABCO).Tetrahydroquinoline product3acould be obtained in good yield with>20:1dr(Table 1,entries 1–3).Next,we evaluated a series of chiral Lewis bases for asymmetric transformation.Chiral tertiary phosphineC1andC2are not effective catalysts,probably due to the steric hindrance.Connon’s chiral PPY catalystC3-C5exhibited high activity,while the products delivered were almost racemic.Using Chen’s PPY catalystC6with an additional shielding group on the prolinol only provided3awith 57:43er.Moderate enantioselectivity was also observed in the reaction with tertiary amineC7.Despite these negative results for the remote stereocontrol,to our gratification,we observed a significant stereorecognition in the reaction usingβ-ICDC8as the catalyst (entry 11).This bifunctional catalyst derived from cinchona alkaloid could provide3ain 65% yield with 88:12er,indicating that properly distributed H-bonds in the transition state may play an essential role in conveying stereochemical information.
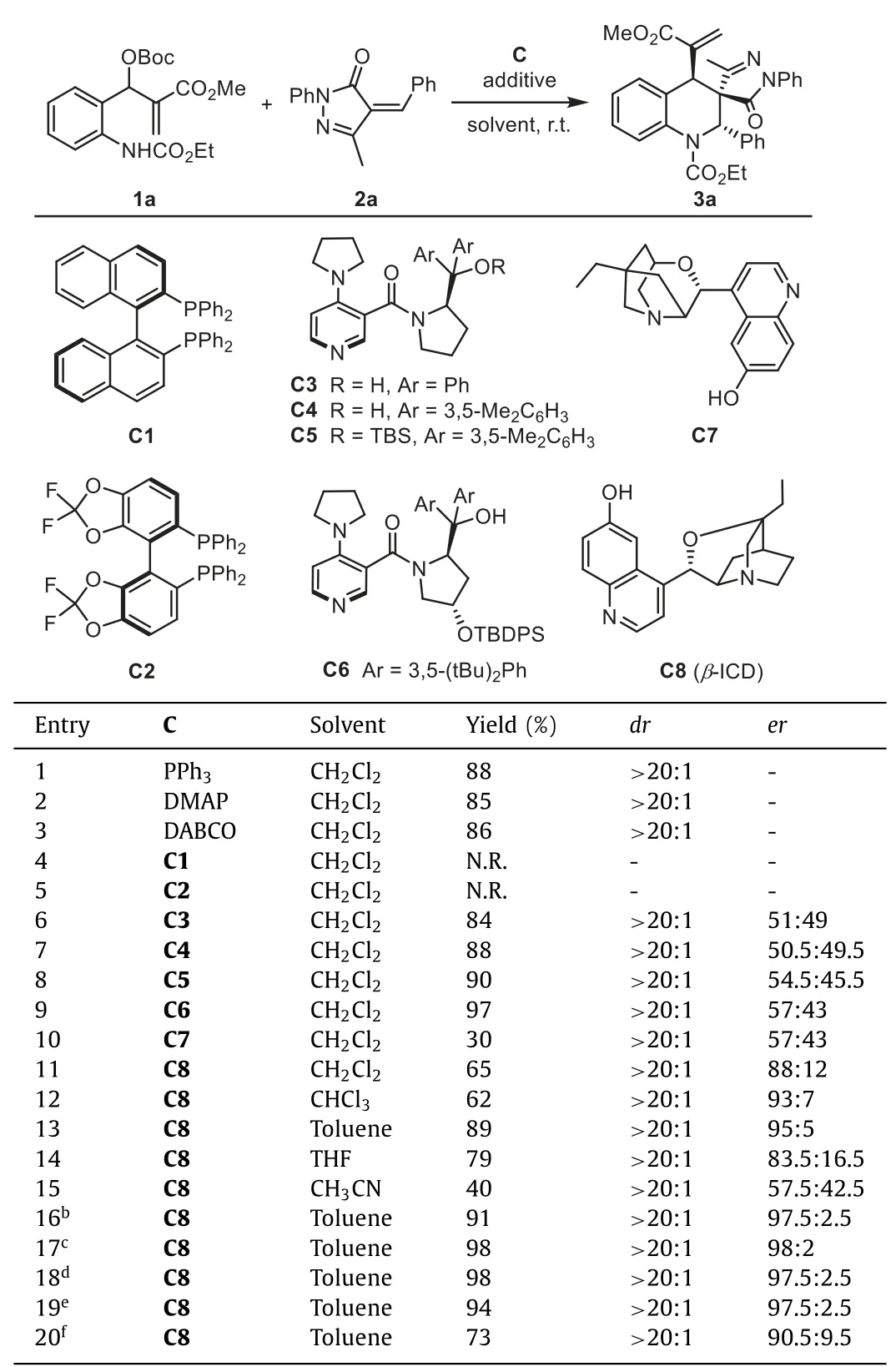
Table 1 Effects of catalyst and solvent on the reaction.a
Further screening of solvents uncovered toluene as the optimum reaction medium for the generation of3a(entry 13,89%yield,with>20:1dr,and 95:5er).The use of ether (THF) or polar solvent (acetonitrile) proved detrimental,resulting in a significant decrease in enantioselectivity (entries 14 and 15).Excellent enantioselectivity could be obtained by adding MgSO4or 4 ˚A MS to the reaction.These results suggest that a trace amount of water in the system may affect the H-bonding betweenC8and the substrate.C8showed high catalytic efficiency and still gave high yields with excellent enantioselectivities at 10% and 5% catalyst loading.When the loading was reduced to 1%,the conversion still could afford3ain 73% yield with a slightly decreased er.Finally,we identified the optimal conditions as listed in entry 19 (5 mol% ofC8,toluene as the solvent,and 4 ˚A MS as the additive at room temperature).
After establishing the simple yet highly effective catalytic system for the remote stereocontrol in the asymmetric cycloaddition of the 1,7-zwitterion,we next explored the substrate scope for the assembly of diverse chiral multifunctionalized tetrahydroquinolines.First,we evaluated the influence of the substituents on theα-phenylidene pyrazolone2.As demonstrated by results summarized in Scheme 2,a range of R1and R2groups are well tolerated,and the stereocontrol of the cycloaddition is not strongly affected by their electronic properties (3a-3m).Electronrich and electron-deficient aryl substituents afford the corresponding spiropyrazolone tetrahydroquinolines in high yields with excellent enantioselectivities (up to 99:1er).The steric properties of R1and R2showed limited influence on the efficiency and stereoselectivity of the reaction.Similar good results could be obtained withortho-substituted (3b),2,4-disubstituted phenyl (3g),and 2-naphthyl (3h) groups.Notably,most cases observed excellent diastereoselectivities,indicating effective stereocontrol at the remote site.
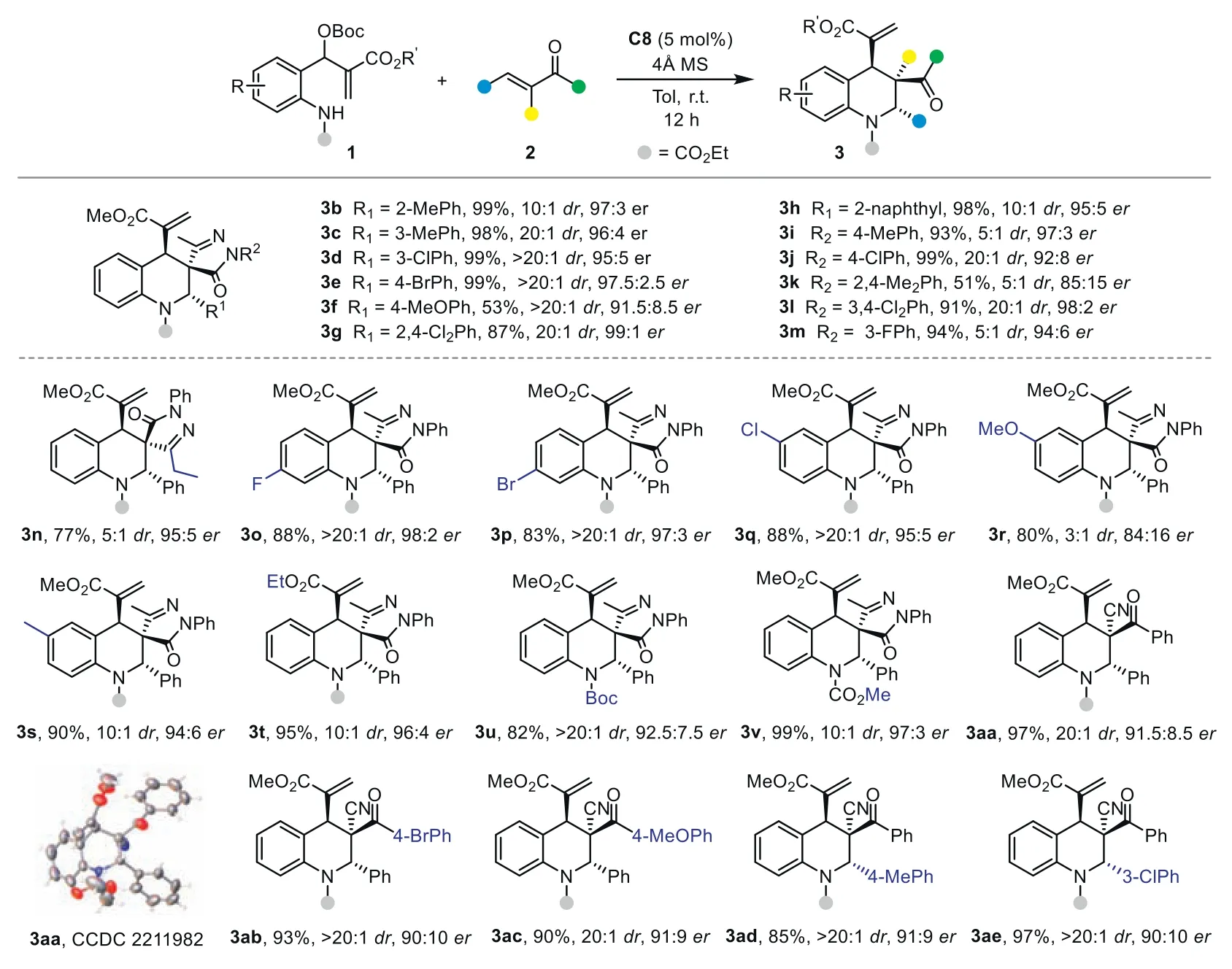
Scheme 2.Substrate scope for the synthesis of multifunctionalized tetrahydroquinolines.Reactions conditions: 1 (0.1 mmol), 2 (0.1 mmol), C8 (5 mol%) in 1.0 mL of toluene for 12 h;isolated yield.
Then,the scope of MBH carbonate1was investigated.Various1bearing different substituents (halogen atom,methyl,or methoxyl group) at different positions of the phenyl ring were compatible in the reaction,providing3o-3sin 83%-90% yields with good stereoselectivities.In addition,MBH carbonate derived from the ethyl acrylate afforded product3tin 95% yield with 96:4er.TheNBoc orN-COOMe-protected substrates gave similar good results (3uand3v).
To further explore the scope of this strategy,we switch to other electron-deficient olefins under identical reaction conditions.The reaction of1awith 2-benzoyl-phenylacrylonitrile smoothly delivered multifunctionalized tetrahydroquinoline3aain 97% yield with exclusive diastereoselectivity and good enantioselectivity.The product structure and absolute configuration of3aawere identified by X-ray analysis.A series of substituted 2-cyano-enones were tested in the process.The reaction efficiency and stereoselectivity were not affected in all these cases (3ab-3ae).Moreover,isopropyl substituted2wand 2-benzylidene-1H-indene-1,3(2H)-dione4proved to be a suitable substrate.The spirocyclic tetrahydroquinoline products3wand5were obtained with good results(Scheme 3a).This further highlights the effective remote stereorecognition and broad scope of our strategy.
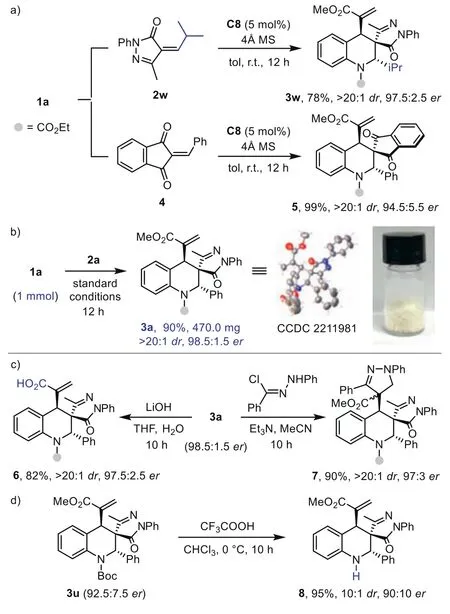
Scheme 3.Other substrates,scale-up synthesis and product derivations.
To demonstrate the practicability of this method,the highly efficient (4+2) cycloaddition reaction was performed on a mmol scale (Scheme 3b).Product3awas produced as a pale-yellow crystalline powder in 90% yield without affecting the stereoselectivities (>20:1dr,98.5:1.5er,Scheme 3b).The structure and absolute configuration of3awere confirmed by X-ray crystallography analysis of (CCDC: 2211981).Remarkably,multifunctionalized tetrahydroquinoline products3containing three consecutive stereocenters could be readily transformed into various derivatives.For example,the ester group of acrylate moiety could be selectively hydrolyzed to afford acid6with highdr,andervalue remained (Scheme 3c).The 1,3-dipolar cycloaddition of3awithin situgenerated azomethine imine smoothly produced derivative7in high yield as a single diastereoisomer.TheN-Boc group could be facilely removed by treating3uwith trifluoroacetic acid (Scheme 3d).
Next,we investigated the reaction mechanism by performing a series of control experiments to elucidate the stereocontrol model.First,caesium carbonate,which could activateo-amino aryl MBH carbonate1aas a Brønsted base,was tested in the reaction with2a(Scheme 4a) [71].Interestingly,no reaction occurred,suggesting that the reaction may not undergo a simple Brønsted base activation pathway.The Pd(0)/L1complex is not an effective catalyst in the process,with only a trace product generated.To further probe the reaction pathway,we conducted a control experiment of2awith9or10.Under the standard conditions,C8could not promote the reaction to afford the (4+2) products,indicating that product3was not formedviaa Brønsted base-catalyzed aza-Michael addition-initiated cyclization.Thus,Lewis base-initiated 1,7-zwitterion cyclization is a more plausible pathway.We then monitored the reaction betweenC8and1aby HRMS analysis.The result indicated that adduct11(m/z: 572.2755) was formedviaSN2′ reaction.Besides key intermediate11and product3a,the generation of intermediate12(m/z: 833.3811) could be detected in the three-component reaction of1a,C8,and2a.These experiments excluded a possible Brønsted base-catalyzed aza-Michael-SN2 process and supported the Lewis base-catalyzed 1,7-zwitterion cyclization pathway well.Based on these results,a possible mechanism was proposed (Fig.1a).Initially,the SN2′ reaction of highly nucleophilic catalystC8with1aafford adduct11.Deprotonation of the allylic site generates 1,7-zwitterionA.The aza-Michael between 1,7-zwitterionAand2adelivers intermediateBand forms the remote stereocenter.Subsequent intramolecular SN2′ reaction leads to the formation of the product and release ofC8.
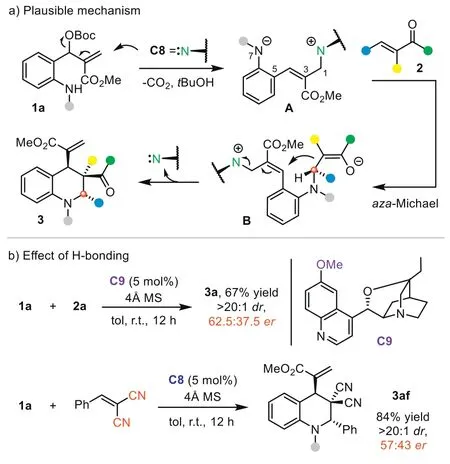
Fig.1.Plausible mechanism and effect of H-bonding.
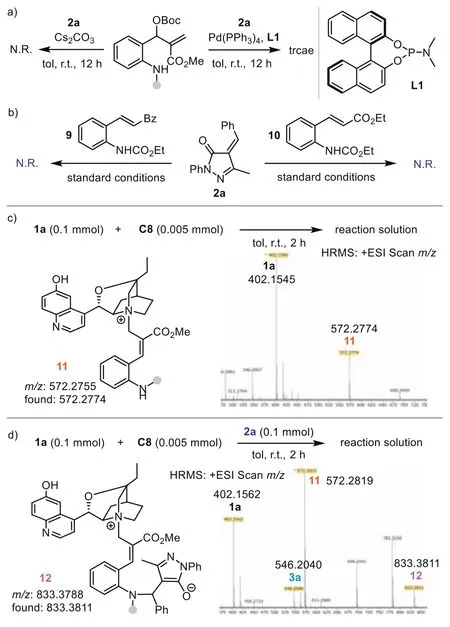
Scheme 4.Mechanistic studies.
Guided by this mechanism,we analyzed the possible transition state for the aza-Michael step,which is crucial for the challenging stereorecognition of the remote prochiral center.To illustrate the possible effect of H-bonding,we tested the reaction usingC9with hydroxyl group methylated.In sharp contrast with the result usingC8(94% yield,>20:1dr,97.5:2.5er),the enantioselectivity of3asignificantly decreased (62.5:37.5er),indicating that the H-bonding between bifunctionalC8and the substrate is the key for longrange stereocontrol (Fig.1b).In addition,the reaction of1awith 2-benzylidene malononitrile gave product3afwith good yield with poorervalue (57:43er),which may result from the non-selective H-bond between each cyano group acceptor with -OH group ofC8.Taking the reaction with2aas an example,we proposed the possible transition state (Fig.2).In the favored TS,the H-bond between the extended quinoline 6-OH group ofC8and carbonyl O-atom of2aguides 1,7-zwitterionAto approach from theSi-face of2a.The aza-Michael addition forms an (S)-stereocenter,finally providing3awith high diastereoselectivity through the induction of adjacent chiral centers.In the disfavored TS,1,7-zwitterionAattacked alkene2afrom theRe-face,while steric repulsion between theNphenyl group of pyrazolone and the cinchona alkaloid scaffold will obviously raise the energy barrier of the aza-Michael step.
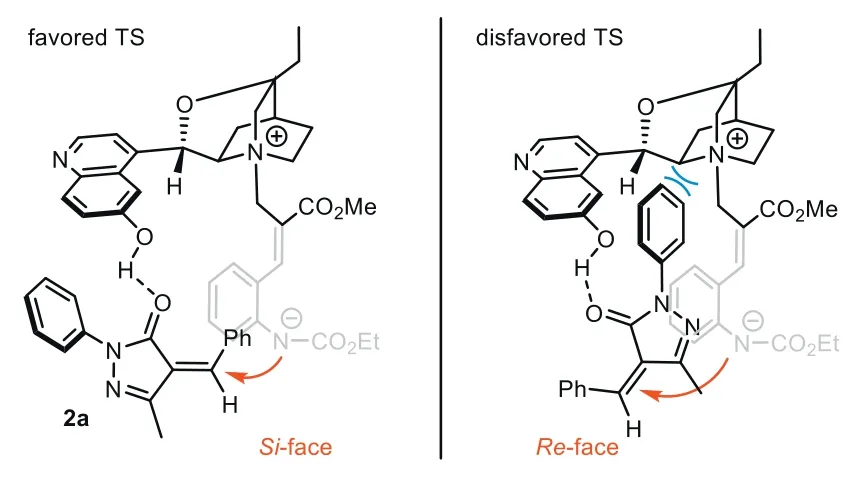
Fig.2.Possible transition states for remote stereocontrol of 1,7-zwitterion.
In summary,we first realized the challenging remote stereocontrol of 1,7-zwitterion intermediates formed from Huang’soamino aryl MBH carbonates.With simple and easily accessibleβ-ICD as the bifunctional catalyst,multifunctionalized tetrahydroquinoline derivatives could be synthesized with excellent enantioselectivity and diastereoselectivity under very mild conditions.The strategy possesses broad substrate scope,and three types of electron-deficient enones are successfully applied under identical conditions.Mechanistic studies disclosed the Lewis base-catalyzed reaction pathway,and H-bonding between the catalyst and enones proves to be crucial for long-range stereocontrol.Moreover,the scale-up reaction and transformations of the multifunctionalized tetrahydroquinoline products demonstrated the potential of this strategy.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
We are grateful for financial support from the National Natural Science Foundation of China (Nos.82073997 and 22001024),the Science &Technology Department of Sichuan Province (Nos.2021YFS0044 and 2021YJ0402),Innovation Team and Talents Cultivation Program of National Administration of Traditional Chinese Medicine (No.ZYYCXTD-D-202209),Xinglin Scholar Research Promotion Project of Chengdu University of TCM.
 Chinese Chemical Letters2024年1期
Chinese Chemical Letters2024年1期
- Chinese Chemical Letters的其它文章
- Spin switching in corrole radical complex
- Benzothiadiazole-based materials for organic solar cells
- Mono-functionalized pillar[n]arenes: Syntheses,host–guest properties and applications✰
- Recent advances in two-step energy transfer light-harvesting systems driven by non-covalent self-assembly✩
- From oxygenated monomers to well-defined low-carbon polymers
- Doping-induced charge transfer in conductive polymers