
苯并三氮唑取代吡啶钴配合物的合成及催化丁二烯聚合行为
2024-04-01 07:04:46郭姜文王书唯张松波胡雁鸣
合成橡胶工业 2024年1期
郭姜文,王书唯*,张松波,胡雁鸣
(1.大连工业大学 纺织与材料工程学院,辽宁 大连 116034; 2.中科院大连化学物理研究所 能源材料部,辽宁 大连 116023)
Brookhart 等在20 世纪90 年代发现α-二亚胺后过渡金属催化剂可高活性地催化烯烃聚合,这一里程碑式的发现为烯烃聚合开辟了崭新的领域[1]。 后过渡金属催化剂具有高极性基团耐受性、配体空间和电子效应,易于调节、活性高是其特点,具有工业化应用潜力[2-3]。然而,后过渡金属催化剂普遍存在热稳定性差的问题,在高温下会快速分解而失活[4]。 基于苯并三氮唑的配体易于制备,可与多种金属配位,在空气和高温下稳定,适合构建多齿配体体系[5]。Duan 等报道了含苯并三氮唑基金催化剂, 用于丙炔Meyer-Schuster 重排和连续丙二烯卤化反应,具有良好的热稳定性和催化活性[6]。 Verma 等合成了含苯并三氮唑的双齿配体,能够有效催化碳碳键、碳氮键和碳硫键耦合反应并对反应物上的功能基团具有良好的耐受性[7]。 本工作合成了一系列苯并三氮唑取代的吡啶基钴配合物,研究助催化剂种类、取代基结构及聚合温度和时间对丁二烯聚合的影响。
1 实验部分
1.1 主要原材料
所有操作均在氮气保护下完成。 四氢呋喃和甲苯经钠/二苯甲酮回流后,在氮气保护下蒸出备用;超干1,4-二氧六环,北京颖诺凯科技有限公司产品;三异丁基铝[Al(i-Bu)3]为1.0 mol/L 的己烷溶液, 上海麦克林生化科技有限公司产品;一氯二乙基铝(AlEt2Cl)为1.0 mol/L 的己烷溶液,阿拉丁试剂(上海)有限公司产品;倍半乙基氯化铝(Al2Et3Cl3) 为0.4 mol/L 的己烷溶液, 研峰科技(北京)有限公司产品;甲基铝氧烷(MAO,1.5mol/L),美国Albemarle 公司产品;1,3-丁二烯,山东鑫圣泰气体有限公司产品。
1.2 仪器分析
核磁共振波谱(NMR)分析采用德国Bruker公司生产的AVANCE Ⅲ400 MHz 型NMR 仪进行,以氘代三氯甲烷为溶剂、四甲基硅烷为内标,于室温下记录NMR 谱图。 液相色谱分析使用美国Agilent 公司生产的Q-TOF 6540 型高分辨飞行时间质谱(ESI-MS)仪记录钴配合物的质谱图。凝胶渗透色谱(GPC)分析采用美国Waters 公司生产的2414 型GPC 仪进行,流动相为四氢呋喃。傅里叶变换红外光谱(FTIR)分析采用日本Shimadzu 公司生产的IRTracer-100型FTIR仪进行。
1.3 聚 合
在手套箱中将称量好的催化剂加入到单口聚合瓶中,密封后取出备用。 将处理好的丁二烯/甲苯溶液加入到聚合瓶中,在设定温度的恒温水浴中预热5 min 后加入助催化剂, 聚合至给定时间后用质量分数1.0%的2,6-二叔丁基-4-甲基苯酚和5%盐酸的乙醇溶液终止反应。 将聚合产物在乙醇中沉淀、洗涤后,在40 ℃真空干燥箱中干燥至恒重。
1.4 配体合成
配体L 1~L 5 按照改进的文献方法制备[8-11]。
L 1 将苯并三氮唑 (10.0 g,63.2 mmol)和2,6-二溴吡啶(15.2 g,126.4 mmol)溶解于50 mL甲苯中并回流18 h, 冷却至室温后倒入200 mL乙酸乙酯中,加入30 mL 质量分数10%的氢氧化钾溶液, 有机相用50 mL 的氢氧化钾溶液洗涤2次,用无水硫酸钠干燥、过滤、浓缩后得到粗产物。目标产物在甲醇中重结晶得到,产率93%。1H-NMR(400 MHz,CDCl3)δ 8.62(dd,J=20.0,6.1 Hz,2 H),8.29(d,J=8.3 Hz,1 H),8.11(d,J=8.3 Hz,1 H),7.96~7.84(m,1 H),7.59(t,J=7.7 Hz,1 H),7.44(t,J=7.6 Hz,1 H),7.30(dd,J=7.2,5.0 Hz,1 H)。13C-NMR(176 MHz,CDCl3) δ 151.1,146.7,140.8,140.0,131.2,129.3,126.1,125.2,119.9,114.7,112.6,77.2,77.0,76.8。
L 2 将苯并三氮唑(0.952 g,8.0 mmol)和2-溴-6-甲基吡啶(0.688 g,4.0 mmol)加入到100 mL的Schlenk 瓶中,混合物在185 ℃下反应40 min。 冷却至室温后加入水和二氯乙烷并用二氯乙烷萃取3 次,将有机相用无水硫酸镁干燥、过滤、浓缩后得到粗产物。 用乙酸乙酯和石油醚作为洗脱剂, 经过层析柱纯化得到目标产物, 产率66%。1H-NMR(400 MHz,CDCl3)δ 8.69(d,J=8.4 Hz,1 H),8.11(dd,J=12.6,8.3 Hz,2 H),7.83(t,J=7.9 Hz,1 H),7.61(t,J=7.6 Hz,1 H),7.46(t,J=7.6 Hz,1 H),7.19(d,J=7.5 Hz,1 H),2.69(s,3 H)。13C-NMR(101 MHz,CDCl3)δ 157.8,151.1,146.6,139.1,131.6,128.7,124.9,121.7,119.7,115.1,111.3,24.2。
L 3 将 苯 并 三 氮 唑(7.14 g,60 mmol)和2,6-二溴吡啶(9.48 g,60 mmol)加入到100 mL Schlenk 瓶中,混合物在180 ℃下反应3 h,冷却至室温后加入100 mL 二氯甲烷,过滤不溶物,去除溶剂得到粗产物,以石油醚和乙酸乙酯作为洗脱液, 经过层析柱纯化得到目标产物, 产率57%。1H-NMR(400 MHz,CDCl3) δ 8.59(d,J=8.4 Hz,1 H),8.27(d,J=8.1 Hz,1 H),8.12(d,J=8.3 Hz,1 H),7.78 (t,J=7.9 Hz,1 H),7.64 (t,J=7.7 Hz,1 H),7.57~7.40(m,2 H)。13C-NMR(101 MHz,CDCl3)δ 151.1,146.8,146.8,140.9,140.0,131.3,129.3,126.2,125.3,119.9,114.7,112.6。
L 4 将L 3(1.65 g,6.0 mmol)、苯硼酸(1.02 g,8.4 mmol)、碳酸铯(3.12 g,9.6 mmol)和四(三苯基膦)钯溶解在40 mL 的1,4-二氧六环中。 混合物回流反应6 h, 冷却至室温后加入水和二氯甲烷并用二氯甲烷萃取3 次。 有机相用无水硫酸镁干燥、过滤、浓缩。 粗产物用石油醚和乙酸乙酯作为洗脱剂, 经过层析柱纯化得到目标产物, 产率83%。1H-NMR(400 MHz,CDCl3)δ 8.79(d,J=8.3 Hz,1 H),8.25(d,J=8.0 Hz,1 H),8.14(dd,J=19.8,8.1 Hz,3 H),8.00(t,J=7.8Hz,1 H),7.76(d,J=7.7 Hz,1 H),7.70~7.61(m,1 H),7.60~7.40(m,4 H)。13C-NMR(101 MHz,CDCl3)δ 156.6,151.5,139.7,138.4,131.7,129.6,129.0,128.8,127.0,124.9,119.8,118.7,114.8,112.7。
L 5 采用L 4 的方法合成L 5, 原料制备时将苯硼酸换为1-萘硼酸(1.44 g,8.4 mmol),产率80%。1H-NMR(400 MHz,CDCl3)δ 8.60(d,J=7.4 Hz,1 H),8.41(d,J=8.3 Hz,1 H),8.31(d,J=8.5 Hz,1 H),8.20~8.05 (m,2 H),8.00 (t,J=8.0 Hz,2 H),7.75(dd,J=7.1,1.1 Hz,1 H),7.65 (dd,J=4.5,3.9 Hz,2 H),7.61~7.36(m,4 H)。13C-NMR(101 MHz,CDCl3)δ 158.2,151.4,146.8,139.4,137.4,134.0,131.6,131.0,129.4,128.9,128.5,127.8,126.6,126.2,125.6,125.4,124.9,123.3,119.7,115.3,112.4。
1.5 钴催化剂合成
钴配合物(Co 1~Co 5)的合成路线如下:
Co 1:在氮气氛围下将氯化钴(0.260 g,2 mmol)和40 mL 四氢呋喃加入到Schlenk 瓶中, 搅拌至完全溶解后滴加配体L 1(0.392 g,2 mmol)的四氢呋喃溶液,在室温下搅拌过夜,将沉淀物过滤后用四氢呋喃洗涤;在40 ℃的真空干燥箱中干燥至恒重,产率43%;ESI-MS:理论值m/z 为522.259 2,测定值为486.0513[M-Cl]+。 Co2:将氯化钴(0.065g,0.5 mmol)和40 mL 甲苯加入到Sschlenk 瓶中,在110 ℃下搅拌1 h,滴加配体L 2(0.252 g,1.2 mmol)的甲苯溶液并回流8 h; 冷却至室温后将沉淀物过滤并用甲苯洗涤,在50 ℃真空干燥箱中干燥至恒重,产率31%;ESI-MS:理论值m/z 为550.313 2,测定值为514.082 6[M-Cl]+。 Co 3 的合成方法与Co 2 相似, 产率29%;ESI-MS: 理论值m/z 为680.051 2,测定值为643.871 8[M-Cl]+。Co 4 的合成方法与Co 2 相似,产率25%;ESI-MS:理论值m/z为674.455 2,测定值为638.116 0[M-Cl]+。 Co 5 的合成方法与Co 2 相似,产率27%;ESI-MS:理论值m/z 为774.575 2,测定值为738.143 6[M-Cl]+。
2 结果与讨论
2.1 取代基类型对聚合的影响
由表1 可知,配体上吡啶2-位的取代基对钴配合物的催化行为影响较大。 随着取代基团空间位阻的增大(见表2),丁二烯聚合速率逐渐降低,聚合物分子量亦随之减小,这是由于取代基空间位阻的增大阻碍了单体与金属中心的配位。 当聚合时间为1 h 时这一趋势表现得更为明显。 取代基为氢时的聚合物收率为91.0%, 为萘基时已降至61.8%。甲基与溴基团具有相近的范德华体积,分别为13.7 cm3/mol 和14.4 cm3/mol,且二者分别为供电子和吸电子基团,由其所得聚合物收率分别为78.0%和74.8%, 这表明取代基的位阻效应对催化行为的影响更为明显。 取代基结构与其聚合物选择性则并无明显的关联规律。 所制备聚合物的分子量分布均较窄[多分散性指数 (PDI)为2.00~2.05],表明基于Co 1~Co 5 的催化体系均具有单活性中心的特点。 鉴于Co 1 表现出高活性、高顺式-1,4-结构选择性及所得聚合物具有高分子量和窄分子量分布的特点,因此采用其研究助催化剂种类及反应温度和时间对聚合的影响。
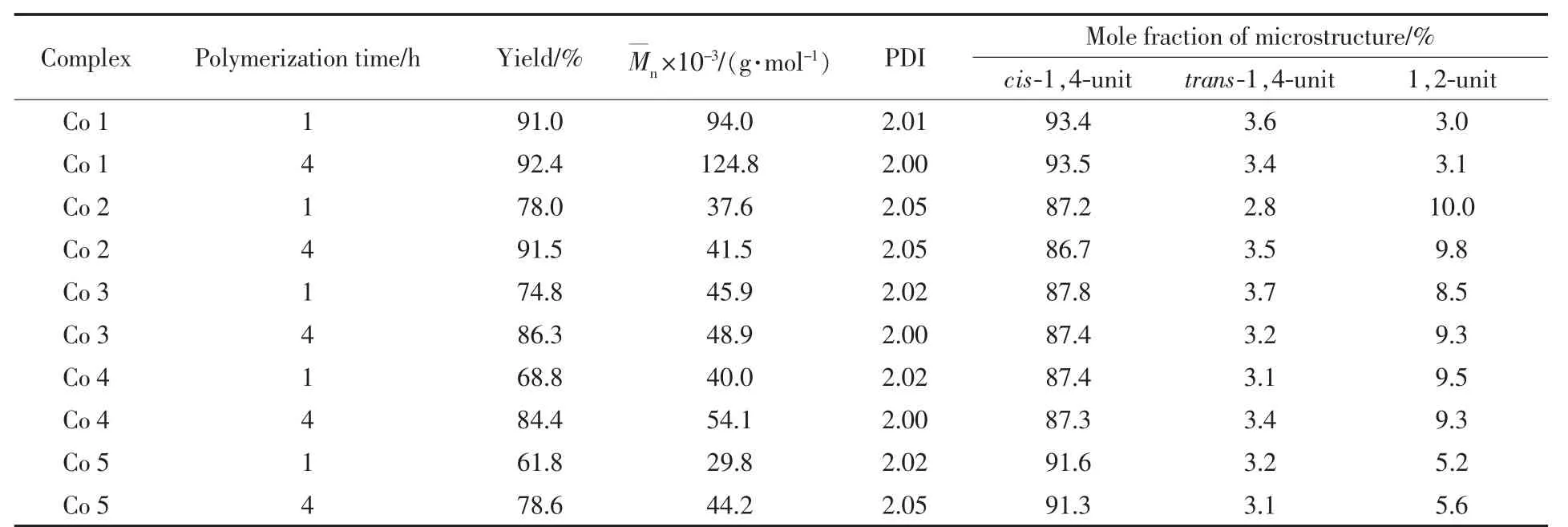
Table 1 Polymerization of 1,3-butadiene with (Co 1-Co 5)/MAO

Table 2 van der Waals volumes of substituents[12]
2.2 助催化剂种类对聚合的影响
以Co 1 为例研究了不同助催化剂对丁二烯聚合的影响,结果见表3。 由表3 可知,以AlEt2Cl为助催化剂时,n(Al)/n(Co)从40 提高到80,催化活性略有下降,聚合产物的顺式-1,4-结构含量和分子量及其分布则基本不变。 采用Al2Et3Cl3为助催化剂时,在n(Al)/n(Co)为5 时,聚合物收率达到99%以上,数均分子量为203.1×103g/mol,但分子量分布较宽(PDI 为4.50),表明体系中存在多种活性中心。 基于Al(i-Bu)3的催化体系对于1,3-丁二烯无聚合活性。以MAO 为助催化剂时,催化体系具有较高的催化活性和顺式-1,4-结构单元(94.0%,摩尔分数,下同),与之前所研究用吡唑取代1,10-菲咯啉氯化钴配合物相当[13]。 聚合产物具有窄分子量分布指数(2.04),且GPC 曲线呈单峰,表明该催化体系具有单活性中心的特征。
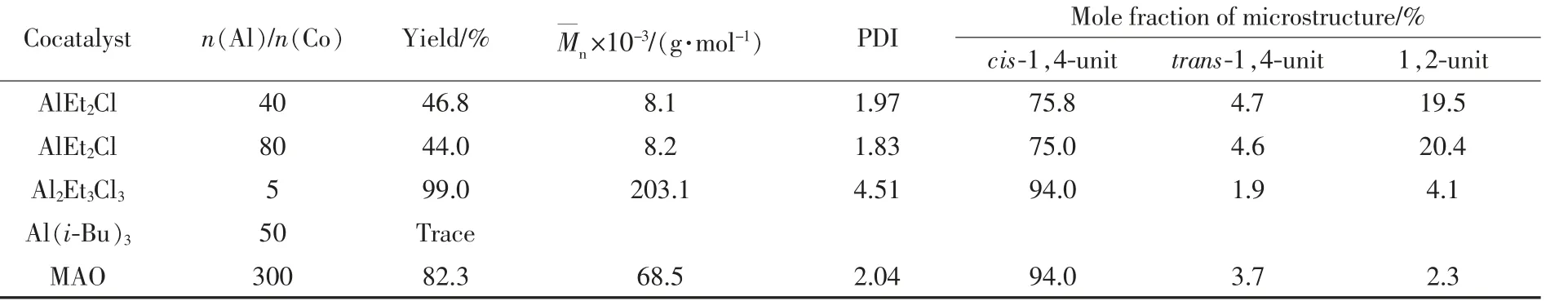
Table 3 Polymerization of 1,3-butadiene with Co 1 activated by different cocatalysts
图1 为聚合产物的1H-NMR 谱图,可以看出化学位移5.37 和5.32 处为1,4-结构的特征峰,4.91 处为1,2-结构的特征峰。根据特征峰积分面积计算得出其1,4-和1,2-结构摩尔分数分别为97.7%和2.3%。 图2 为同一聚合物的FTIR 谱图,其中738 cm-1、911 cm-1和967 cm-1处分别为顺式-1,4-、1,2-和反式-1,4-结构的特征峰, 根据文献[14]中的公式计算得到其摩尔分数分别为94.0%、3.7%和2.3%。

Fig 1 1H-NMR spectrum of polybutadiene activated by MAO
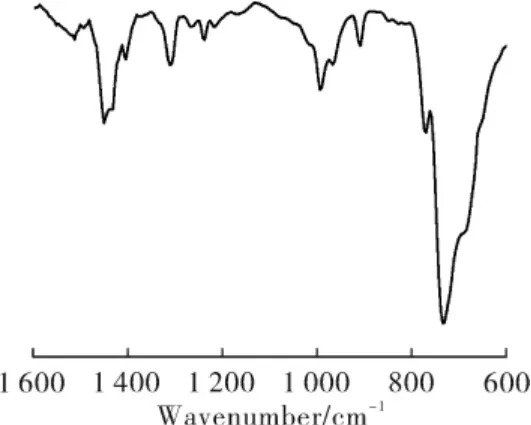
Fig 2 FTIR spectrum of polybutadiene activated by MAO
2.3 反应温度和时间对聚合的影响
后过渡金属催化剂普遍存在热稳定性较差的问题, 当反应温度过高时会导致催化剂分解。用Co 1/MAO 体系研究了反应温度对丁二烯聚合的影响(见表4)。由表4 可知,升高温度可显著提高聚合反应速率, 在0 ℃下反应4 h 的聚合物收率为57.5%,在80 ℃反应1 h 聚合物收率即高达93.6%, 这也表明苯并三氮唑取代吡啶钴催化剂具有较好的热稳定性。 反应温度在0~50 ℃时,聚合物的顺式-1,4-结构含量基本不变(93.2%~94.0%),其PDI 均保持在2.0 左右;进一步升高温度至80 ℃,由于链转移反应加剧导致聚合物分子量降低,但分子量分布仅略有加宽;同时,聚合物顺式-1,4-结构含量降低,反式-1,4-和1,2-结构含量升高,这是因为在高温下,对式烯丙基结构活性中心向热力学更稳定的同式结构转变所致[15]。
3 结 论
a)以苯并三氮唑取代吡啶为配体的氯化钴配合物, 在MAO 活化下催化丁二烯聚合具有较高的催化活性和热稳定性,且具有单活性中心的特点。 所得聚合物具有高顺式-1,4-结构含量(94.0%)和窄分子量分布特征(PDI 为2.0)。
b)配体结构对配合物的催化行为影响较大,聚合活性随配体上吡啶2-位取代基空间位阻的增大而降低, 取代基结构与其聚合物顺式-1,4-结构选择性并无明显关联。
c)升高反应温度可显著提高聚合速率,聚合物分子量降低,PDI 仅略有增大,基本维持在2.0。

Table 4 Polymerization of 1,3-butadiene with Co 1/MAO system at different polymerization temperatures
猜你喜欢
中国特种设备安全(2022年1期)2022-04-26 14:16:10
云南化工(2021年10期)2021-12-21 07:33:28
纺织科学研究(2021年7期)2021-08-14 01:42:30
化工学报(2020年4期)2020-05-28 09:25:24
今日农业(2019年11期)2019-08-13 00:49:02
化工管理(2017年18期)2017-03-03 16:40:34
化工设计通讯(2017年9期)2017-03-02 16:22:38
现代检验医学杂志(2016年1期)2016-11-12 13:19:54
国外医药(抗生素分册)(2016年4期)2016-07-12 14:25:21
当代化工研究(2016年2期)2016-03-20 16:21:20
