
基于化学环境自适应学习的掺杂石墨氮化碳纳米片光学带隙预测
2024-03-22 03:05陈宸张继勇侯佳
中国传媒大学学报(自然科学版) 2024年1期
陈宸,张继勇,侯佳
(1.杭州电子科技大学,杭州 310037;2.杭州电子科技大学丽水研究院,丽水 323060)
1 引言
目前,各种新型的疾病陆续出现,例如几年前出现的大规模新冠病毒肺炎。这些情况需要的是新型的特效药,但药物研发成本极高始终让制药行业面临许多挑战和困难。
主要的挑战有以下两点,第一,新药研发的时间成本很高,一种新药物的研发基本都超过了十年;第二,新药研发所需要的经济成本投入也很高,其平均投入的成本高达25亿美元,这主要是因为药物需要在临床实验下才能测验,成功率和回报率很低。在此过程中,1/3 的时间和金钱都投在了药物发现的早期阶段。因此,缩减药物发现的时间以及扩大药物数据库成了必要的环节之一。开发出有效的自动化技术,在广阔而离散的化学空间中快速发现可能的、多样化的候选分子,对制药行业来说很有吸引力。
石墨氮化碳(g-C3N4)[1]自1834 年由Berzelius 和Liebig[2]发现后不断地发展,已经在能源、环境、电子和生物医学等多个领域得到广泛应用。2009年Wang等[3]发现并证实了g-C3N4非凡的光催化性能,因为石墨氮化碳的2.7eⅤ带隙结构使其对可见光敏感[4],所以可以利用太阳光能将水分解成氢气和氧气,极大提升了水分解的效率。不仅在水分解方面,研究人员在CO2 还原、有机废水处理等方面也进行了深入研究[5-9]。改进光催化活性、提高稳定性和理解光催化机理是当前研究的焦点。
图结构数据因其特殊的结构,广泛存在于各个领域,例如社交网络、生物信息学、化学分子分析等。图神经网络(GNNs,Graph Neural Networks)作为专门处理图结构数据的深度学习模型,其早在2005年便拥有了雏形,这个时期的方法都是基于手工涉及的特征和规则,直到2016 年图卷积网络(GCN,Graph Convolutional Network)[10]的问世。图神经网络另一个重要的基石便是消息传递神经网络(MPNN,Message Passing Neural Network)[11]。 MPNN 的概念最早由Justin Gilmer等人在2017年提出,是一种用于量子化学问题的消息传递框架。图卷积网络和消息传递神经网络是专门设计用于学习图表示以进行下游预测任务(包括分类和回归)的流行深度学习模型。GCN 已在分子动力学[12]和医学疾病[13,14]的数据分析中得到广泛应用,而MPNN 推广了多类空间GCN 以学习分子特征[15-17],并在分子性质预测中展现了良好的结果[18-22]。这两种方法都依赖于从节点及其邻居那里聚合信息以生成节点表示。在MPNN中,消息通过消息函数在相邻节点之间传递,然后通过顶点函数更新节点嵌入,最终通过从图中节点嵌入中读取函数生成分子特征表示。
图神经网络与底层化学键拓扑结构的概念相似性促使了广泛应用GNNs来预测量子力学性质。例如,在晶体图卷积神经网络(CGCNN,Crystal Graph Convolutional Neural Network)[23]的研究中,成功地预测了46744个晶体的八个物理性质,如能隙和费米能级。图同构网络(GIN,Graph Isomorphism Network)[24]被设计用于离散特征空间,以在实际应用中区分同构图结构。最近,神经等变原子间势(NequIP,Neural Equivariant Interatomic Potentials)[25]的开发使其能够以密度泛函理论的准确度进行分子动力学模拟,同时仅需其计算成本的一小部分。
除了上述几种针对量子力学领域的图神经网络外,经典图论中还有GraphSAGE(Graph Sample and Aggregation)[26]、GAT(Graph Attention Network)[27]等图神经网络模型。2017 年Hamilton 等[26]提出了GraphSAGE,其核心在于通过采样每个节点的局部邻域信息构成子图,并将这些子图通过聚合的方式来捕捉节点的信息和全图结构信息。GAT 由Ⅴeličković等[27]在2018 年提出,其核心在于引入了注意力机制,简单来说就是通过计算每个节点与其邻居之间的的注意力权重来判断节点的重要性,以此来动态地分配节点权重,使模型可以更灵活地捕捉节点之间的复杂关系,提高了模型的表达能力。主邻域聚合(PNA,Principal Neighbourhood Aggregation)[28]引入了不同的缩放器,用于控制中心节点的聚合消息的权重。Deepergcn[29]提出了一个由连续变量参数化的广义聚合函数。通过学习变量,广义聚合函数可以被视为Mean和Max 的组合。这两种方法都是使用了多种聚合器进行消息的聚合,当前拥有良好的效果。如图1所示,不同分子结构分别在四种聚合器情况下的差异,这就可能出现在使用Mean聚合器时两个分子结构拥有几乎相同的结构,但使用别的聚合器时却差异巨大,为了解决这个问题,本文提出了基于化学环境的图神经网络,本文的模型相比于其他模型拥有良好的效果,原因就是这些分子的性质在很大程度上依赖于局部原子间的相互作用,也就是分子本身的结构特征,而本文模型可以更好地捕捉分子结构的信息。
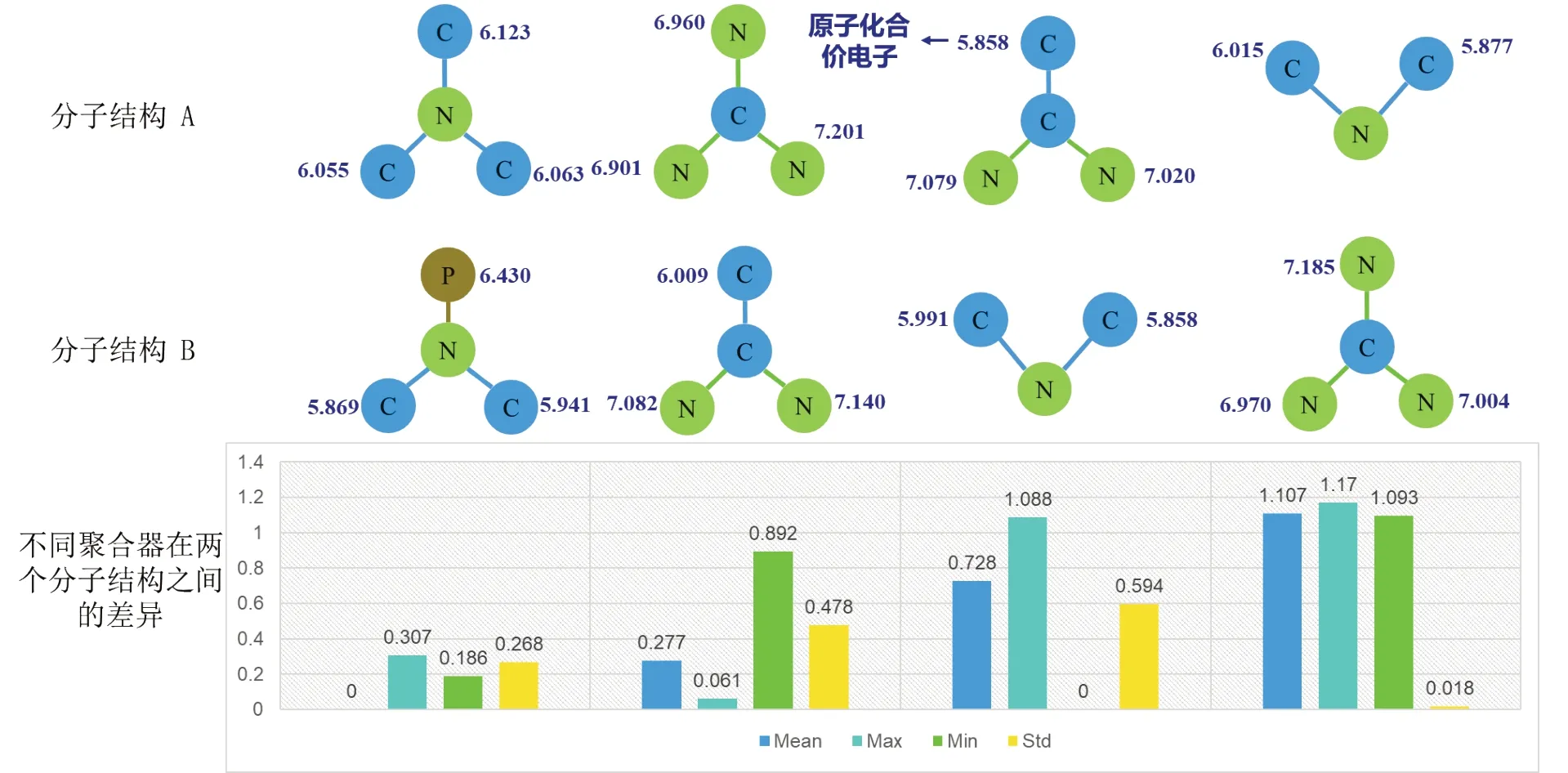
图1 单一聚合器问题讨论
2 化学环境图神经网络
基于化学环境的图神经网络为了用于预测g-C3N4纳米片及其掺杂变体光学带隙的整体框架如图2所示。该模型将分子结构作为输入,并预测分子的重要属性之一光学带隙。首先将分子结构视为一张图,原子被解释为图中的节点,化学键被解释为边,使用G(V,E)来表示。边缘特征首先通过边缘编码器进行线性转换,使其具有与节点特征向量相同的大小。一系列隐藏层被利用来聚合来自相邻节点的信息,以提取潜在的节点嵌入,而这里的隐藏层就是CEAL(Chemical Environment Adaptive Learning)层,顾名思义就是化学环境自适应学习层,也是本文提出模型的核心。由最后一个CEAL 层提取的节点嵌入被发送到一个读出层,以形成图级表示,其中的读出层拥有多种选择,本文中选择了求和池化层(Add Pooling)。最后,图表示被用来预测g-C3N4 及其掺杂变体的光学带隙。
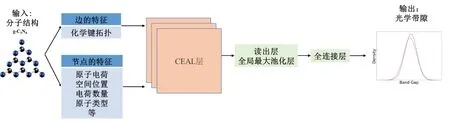
图2 基于化学环境的图神经网络模型整体结构
2.1 自适应聚合机制
为了有效地学习原子相互作用的表示,我们将可学习的权重应用于聚合器。这种方法允许根据模型的学习效果来自适应分配聚合器的权重,并使模型能够捕捉原子相互作用的变化,从而改进了分子性质预测的效果。这些权重在训练过程中自适应地学习,以确定基于原子的局部化学环境的最佳聚合函数组合。通过为有限的聚合器集合分配可学习的权重,并制定它们的任意线性组合来实现这一点,写成公式如下:
其中i和j表示中心节点和邻居节点表示在第k层时中心节点接收到的消息是在第(k-1)行时的节点特征,⊕A是多个聚合器的组合是在第k层时可学习的聚合器权重。经过多次迭代后的最终聚合消息和中心节点的特征共同输入到中,更新中心节点嵌入的公式如下所示:
2.2 聚合器
表格1 中展示了所有本文所涉及的聚合器,上文中提到的⊕A就是聚合器的排列组合,也就是A ={}Sum,Meɑn,Mɑx,Min,Std, 其中包含了多样化的聚合函数,不仅是只有展示的五个聚合器,以此来提取原子化学环境的各种特征。候选函数集包含具有足够多样性的聚合函数,希望在将来能够加入更多的聚合器,以扩展搜索空间并且提高模型性能。
除了这些常用的聚合器外,模型支持加入更多聚合器来提升消息的掌握程度,由于不同的聚合器拥有不同的效果,因此希望加入更多的聚合器来加入更多的处理消息方式。例如归一化矩阵聚合器,也就是Skewu(X k)和Kuru(X k)聚合器,这是一种基于n值的归一化表示,其中当n= 3时表示偏度,n= 4时表示峰度。这通常通过计算归一化的三阶矩来衡量,偏度为正表示分布右偏,为负表示左偏,为零表示对称;峰度通常通过计算归一化的四阶矩来衡量,峰度的不同值表示分布尾部形状的不同特征,如尖峭或平坦。这些归一化矩聚合函数的引入旨在通过考虑更高阶的统计信息来捕捉节点邻域的更复杂特征。偏度和峰度聚合器的表达如下所示:
一组聚合器(下表1中有详细说明)被利用来收集节点的化学环境的各个方面的属性,如同图3中所展示的,均值聚合器Mean可以捕获分子属性中的电子密度属性,通常用来描述原子、分子或晶体中电子的分布情况,对理解化学键和分子结构等方面非常重要;最大值聚合器Max可以获得分子属性中的电负性属性,是描述一个原子在化学键中吸引和保留电子的能力的物理性质,通常用来衡量一个元素或原子对共价键中电子的相对亲和力,常用于解释分子和化合物的性质,特别是在预测共价键的性质和极性方面起到关键作用;标准差聚合器Std可以收集到来自邻居的电极性,通常用来描述电场中的正负电荷分布或电路中的正负电极,涉及到正电荷和负电荷之间的相互作用和分布;偏度聚合器Skew可以通过计算偏度获得邻居的电四极矩,是描述电荷分布不对称性的物理量,是电多极矩中的第二项,表示电荷分布在三维空间中的非均匀性,与电偶极矩和电单极矩一同描述了物体的电性质,在描述分子或原子的电场分布时有重要作用。

表1 节点u在第k层的相应聚合消息表达
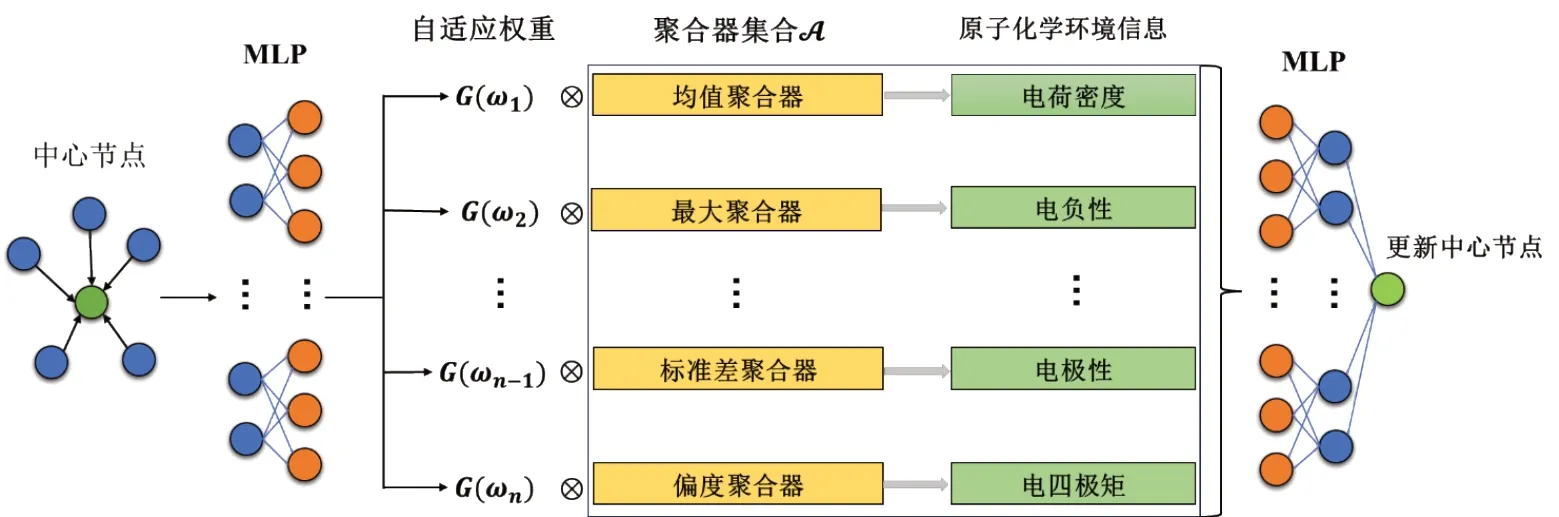
图3 CEAL层的架构示意图
3 实验结果分析
3.1 数据集和实验环境
首先介绍核心数据集石墨氮化碳g-C3N4,数据集拥有g-C3N4本身以及8个掺杂变体,总共拥有大约110000个分子,每个分子由126个碳、氮等原子组成,形成所需要的大规模晶体状分子。该数据的所有分子都通过AIMD在1纳秒内捕捉其轨迹,其中所包含的信息拥有空间坐标、原子类型和角动量通道上的电子数量等,而AIMD均使用开源CP2K[30]软件进行,该软件采用Goedecker-Teter-Hutter(GTH)赝势方法[31]、Heyd-Scuseria-Ernzerhof(HSE06)交换相关泛函[32]和极化价-双-ζ(PⅤDZ)基组[33]。通过选择的HSE06范围分离的混合泛函得到了优化的未掺杂g-C3N4纳米片,验证得到其计算带隙为2.78电子伏特。此外,对选择的每个原子构型,还计算了其角动量分辨的Mulliken电荷[34],因为这与决定带隙的电子分布直接相关。因此,原子坐标和角动量分辨的Mulliken电荷被用作我们的机器学习模型的输入数据,以预测未掺杂和掺杂的g-C3N4纳米片的带隙;其次是QM9数据集,其中报告计算了由CHONF组成的13万有机小分子,其数据来自于GDB(Gaussian Database),包含关于每个分子的许多属性,包括电子结构属性(HOMO、LUMO能级等)、热力学性质(能量、熵、自由能等)等,本文选择了一部分经典的分子作为研究对象;最后是FePt数据集,其是通过OLCF Constellation提供的FePt合金数据集,其化学属性包括总焓、原子电荷转移和原子磁矩等,每个原子样本都具有2 × 2 × 4的体心立方结构的超晶胞,该数据集是使用一种局部自洽多重散射(DFT,Density Functional Theory)应用程序计算得到。无掺杂g-C3N4纳米片及其八种掺杂变体的光学带隙数据集拥有大约110,000个原子,都是通过AIMD 模拟获得的。这些数据集包括了未掺杂的也就是原始的g-C3N4,而掺杂变体的命名在上文中描述过,例如C1P等,也就是将图4中C1的原子替换成P原子,剩余的数据集也是相同的命名方法。

图4 g-C3N4分子结构图
3.2 对比试验分析
为了展示基于化学环境的图神经网络在准确预测具有多样分类结构的分子的光学带隙方面的优越能力,模型使用了包含g-C3N4 及其八种掺杂变体的所有九个数据集进行训练。本文测试了一系列经典图论和本文模型进行比较,结果如图5所示,可以发现本文模型在预测的准确度上有明显的提升,平均绝对误差达到了0.031 eⅤ,并且在每个模型上进行了五次的实验,模型的波动也明显较小,拥有更好的稳定性。
此外,如图6所示,模型对光学带隙的预测结果点更加集中在对角线上,表明本文模型在预测准确性上优于其他经典图论中的GNN 模型。所有这些结果都充分肯定了本文模型准确地从原子的化学环境中提取信息的显著能力,以将分子结构映射到光学带隙。重要的是,这种有效提取局部化学环境信息的能力将使得能够准确预测任何受原子相互作用影响的其他分子性质,加速了对具有所需功能的新材料的实验发现。实验结果表明,所提出的基于化学环境的图神经网络模型在光学带隙预测的平均绝对误差方面表现出了一致的优越性能。如表2 所示,本文定量测试了g-C3N4的九个单独数据集在整体模型上的效果,可以发现不论是哪种形式的掺杂变体,基于化学环境的图神经网络都拥有良好的效果,平均绝对误差都保持在0.03 eⅤ上下波动,说明模型在预测大部分分子的光学带隙时拥有良好的准确率和极小的误差。
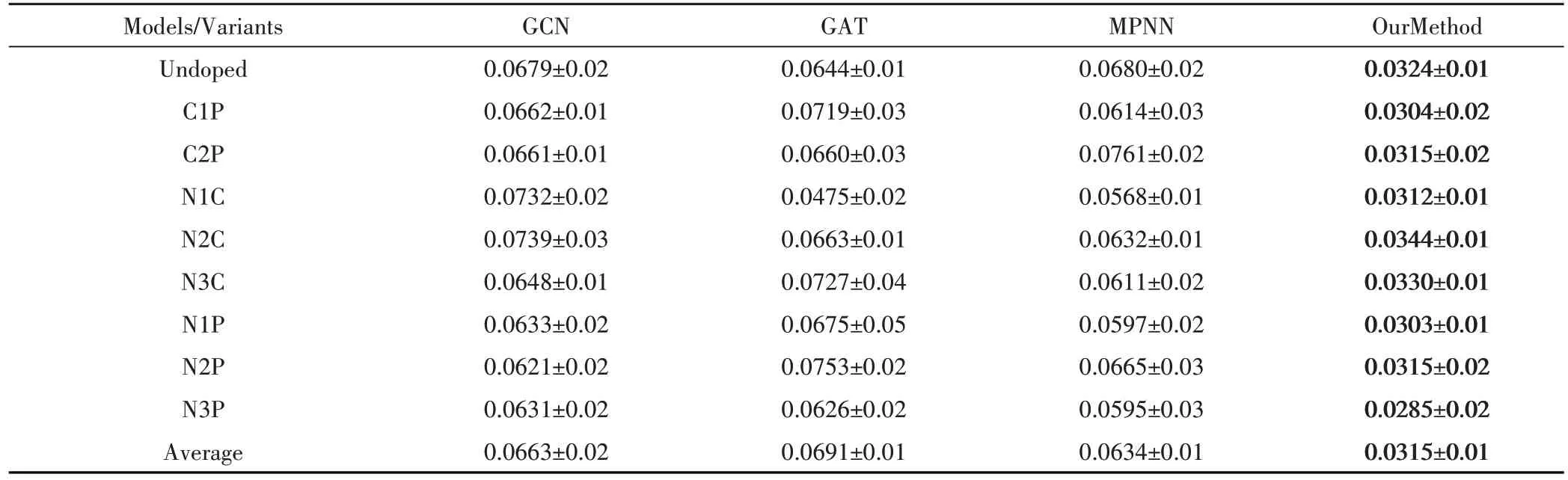
表2 g-C3N4的九个单独数据集在整体模型上的效果

图6 各个图神经网络的散点图结果
4 结论
本文针对使用单一聚合器的问题进行讨论,提出了基于化学环境的图神经网络并利用自适应聚合机制从原子的化学环境中提取深层见解,解决了单一聚合的局限性。实验结果表明,所提出的模型可以显著提高石墨氮化碳纳米片及其掺杂变体的光学带隙预测能力,其在各项指标上都优于其他方法。此外,分子性质预测的目的是从分子化合物的分子结构来预测其各种物理和化学性质。这项任务对药物发现、材料设计和环境风险评估至关重要,因为它可以深入了解化合物的性质,而不需要昂贵和耗时的实验测量。
猜你喜欢
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
农村青少年科学探究(2020年5期)2020-08-18
中学生数理化(高中版.高考理化)(2019年6期)2019-06-22
新民周刊(2018年8期)2018-03-02
饮食科学(2017年12期)2018-01-02
中学化学(2016年10期)2017-01-07
湖南城市学院学报(自然科学版)(2016年2期)2016-12-01
少儿科学周刊·少年版(2015年1期)2015-07-07
