
利用CRISPR/Cas9 技术构建Quaking 敲除的小鼠胚胎成纤维细胞株
2024-03-12 14:31高登科马白荣郭怡莹刘薇刘田靳亚平江舟陈华涛
生物技术通报 2024年2期
高登科 马白荣 郭怡莹 刘薇 刘田 靳亚平 江舟陈华涛
(1.西北农林科技大学动物医学院,杨凌 712100;2.西北农林科技大学 农业农村部动物生物技术重点实验室,杨凌 712100;3.四川大学国家卫生健康委员会时间生物学重点实验室,成都 610000)
Quaking(QKI)蛋白属于进化保守的含有核不均一核糖核酸蛋白K 同源结构域(K homology,KH)的RNA 结合蛋白STAR(single transduction and activation of RNA,STAR)家族成员,因Quaking 基因缺失导致中枢和外周神经系统出现神经髓鞘发育障碍的现象,小鼠机体表现为颤抖,故将引起此现象的基因命名为Quaking[1]。QKI 的3 种亚型QKI-5、QKI-6 和QKI-7 拥有相同的KH 结构域,主要区别在于C 端的30 个氨基酸不同[2]。QKI-5 中包含一个核定位信号,可以在细胞核和细胞质之间穿梭,主要在细胞核中表达;QKI-6 可以在细胞核和细胞质中表达,而QKI-7 主要分布在细胞质中[3]。
QKI 是影响RNA 调控的关键调节因子,其能与RNA 特异性结合,在pre-mRNA 的剪接、microRNA的调控与环状RNA 的形成等方面发挥重要作用[4]。QKI 可作为一种肿瘤抑制因子抑制肿瘤的发生。有研究发现,QKI-5 在良性前列腺增生组织中高表达,而在癌组织中低表达,并且QKI-5 可有效抑制前列腺癌细胞的增殖[5]。同时,有研究发现在乳腺癌细胞中QKI-5 的高表达可抑制癌细胞增殖[6]。此外,特异性过表达QKI-5 能抑制卵巢癌细胞A2780的增殖,并促进癌细胞发生凋亡[7]。在免疫调控中,QKI 是参与单核细胞或巨噬细胞分化和功能调节的关键因子。QKI 通过调节Ahr/STAT1-NF-κB 途径,在抑制先天免疫反应方面发挥重要作用[8]。在神经系统中,QKI-5、QKI-6 与QKI-7 蛋白在髓鞘形成细胞和星形胶质细胞中均有显著表达;QKI 作为髓鞘形成的调节因子,是髓鞘形成的关键蛋白[9]。QKI 作为一种RNA 结合蛋白,是少突胶质细胞中核mRNA 输出的调节因子,并参与了髓鞘形成[10]。QKI 在血管发育中起着至关重要的作用,Quaking 突变导致的血管缺陷会引起胚胎死亡[11]。QKI 也是血管生成的正调节因子,其细胞作用是调节内脏的内胚层功能,包括维甲酸的局部合成以及随后控制内皮细胞增殖、基质产生和内脏内胚层存活,这对于血管重塑至关重要[12]。有研究证据表明,QKI 可能通过参与维持肌卫星细胞的储备,促进肌肉细胞进入静息期,抑制小鼠成肌细胞的分化[13]。综上所述,Quaking 基因编码的RNA 结合蛋白QKI 在癌症发生、免疫调控、髓鞘形成、血管生成和肌肉发育等生理过程中发挥重要作用,然而QKI 在小鼠胚胎成纤维细胞(NIH3T3)中的生物学功能目前尚不清楚。
1 材料与方法
1.1 材料
限制性核酸内切酶FastDigest Esp3I、TurboFect转染试剂、Donkey anti-Rabbit IgG(H+L)Highly Cross-Adsorbed Secondary Antibody(Alexa Fluor 488)均购于美国Thermo Fisher 公司;T4 DNA Ligase 购于日本TaKaRa 公司;DH5α 感受态细胞购自北京天根生化科技有限公司;HEK293T 细胞和NIH3T3 细胞购买于中国科学院典型培养物保藏委员会细胞库;胎牛血清购自美国Gibco 公司;DMEM 高糖培养基购自Hyclone 公司;胰蛋白酶消化液、嘌呤霉素、Polybrene、RIPA 裂解液、DAPI 染色液均购于碧云天公司;pcDNA3.1-Quaking 过表达质粒为实验室保存[14];LentiCRISPRv2 载体、辅助质 粒psPAX2 和pMD2.G 质粒由西北农林科技大学动物医学院萧飒教授馈赠;BCA 蛋白浓度测定试剂盒购自江苏凯基生物技术股份有限公司;脱脂奶粉购于美国BD 公司;HRP 共轭山羊抗兔抗体购于中国中杉金桥公司;兔抗GAPDH 抗体购于武汉三鹰生物技术有限公司;兔抗QKI 抗体购于博士德生物工程有限公司;ECL化学发光液购自北京迪宁生物科技有限公司;Trition X-100、BSA 试剂均购于Sigma 公司;CCK8 试剂购于米鼠生物公司。
1.2 方法
1.2.1 设计并合成sgRNA 在NCBI 数据库中获取小鼠Quaking 基因的基因组序列信息;利用CRISPR在线设计网站(http://crispor.tefor.net/)提交Quaking基因的外显子核苷酸序列,选取系统自动生成的评分最优的2 组sgRNA,分别命名为sgRNA1、sgRNA2;选择Cas9 质粒中所需的慢病毒载体LentiCRISPRv2,生成含有Esp3I 酶切位点的sgRNA序列[15],交由西安擎科泽西生物有限公司合成,设计的序列信息见表1。
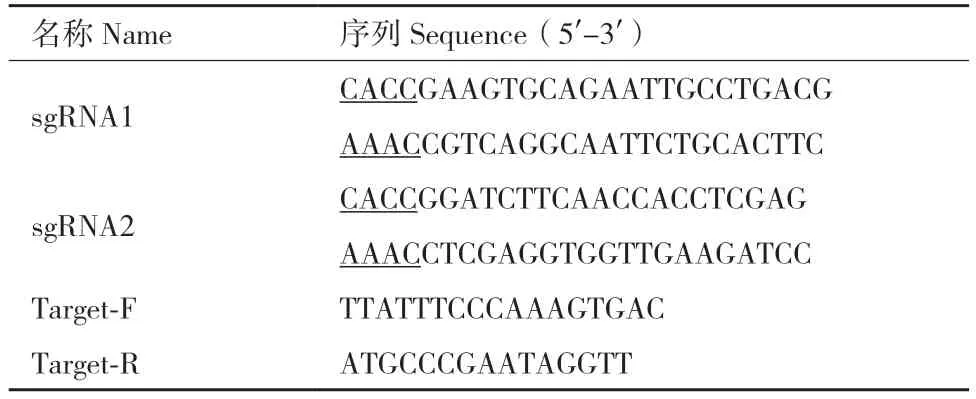
表1 序列信息Table 1 Sequences information
1.2.2 构建sgRNA 敲除载体 将互补的sgRNA寡核苷酸单链退火形成双链。使用限制性核酸内切酶Esp3I 酶切慢病毒载体LentiCRISPRv2,以获得线性化载体。sgRNA 退火产物与线性化载体LentiCRISPRv2 使用T4 DNA Ligase 连接过夜。随后将连接产物转化至大肠杆菌DH5α 感受态细胞中,取适量培养后的转化体系菌液涂布平板观察菌落生长状况,挑取单克隆菌落培养16 h。培养后的菌液提取质粒并送西安擎科泽西生物有限公司进行测序,将测序验证正确的重组质粒命名为LentiCRISPRv2-sgRNA1、LentiCRISPRv2-sgRNA2。
1.2.3 筛选敲除效率高的重组慢病毒质粒 HEK-293T 细胞用含10%胎牛血清的DMEM 高糖培养基培养,细胞密度达到70%-80%时,利用TurboFect转染试剂进行转染。转染4 组,转染质粒分别为:pcDNA3.1-Quaking;pcDNA3.1-Quaking+LentiCRISPRv2;pcDNA3.1-Quaking+LentiCRISPRv2-sgRNA1;pcDNA3.1-Quaking+LentiCRISPRv2-sgRNA2。在37℃培养箱培养48 h 后收集细胞样品,Western blot 检测QKI 蛋白的敲除效率,以筛选获得敲除效率高的重组慢病毒质粒。
1.2.4 嘌呤霉素最低致死浓度筛选 NIH3T3 细胞用含10%胎牛血清的DMEM 高糖培养基培养,选择生长状况良好的NIH3T3 细胞,经胰蛋白酶消化后加入等体积含10%胎牛血清的DMEM 高糖培养基终止消化,计数后调整细胞悬液浓度至5 × 104个/mL,96 孔板中每孔加100 μL 细胞悬液,待细胞贴壁后加入0-5 μg/mL 不同梯度浓度的嘌呤霉素,每0.5 μg/mL 设置一个浓度梯度,每个浓度设置6 个重复。观察细胞状态,将加药后48 h 内细胞全部死亡的最小浓度作为NIH3T3 细胞的嘌呤霉素最低致死浓度,此浓度用于后续阳性克隆细胞株的筛选。
1.2.5 慢病毒包装及转导NIH3T3 细胞 将HEK293T 细胞铺板于6 孔细胞培养板,待细胞生长至密度为70%-80%时,把筛选获得的敲除效率高的重组慢病毒质粒和辅助质粒psPAX2、pMD2.G 按照一定比例(2∶1∶1)共转染至HEK293T 细胞。转染16 h 后,将培养液更换为新的含10%胎牛血清的DMEM 高糖培养基,继续培养48 h 后收集细胞上清液,3 000 r/min 离心5 min 取上清即为病毒原液,0.45 μm 滤器过滤、分装后置于-80℃保存备用。提前1 d 铺好状态良好的NIH3T3 细胞,将收集的慢病毒液与1 mL 无血清DMEM 高糖培养基混匀,并加入终浓度为10 μg/mL 的Polybrene 以提高感染效率。弃去旧培养基,用PBS 清洗一遍后加入毒液,感染48 h 后加入嘌呤霉素进行抗性单克隆细胞的筛选,将单克隆细胞扩大培养后,收取细胞样品提取基因组DNA,使用表1 中的Target 引物进行PCR 扩增后测序鉴定并比对结果,随后采用Western blot 对单克隆细胞株中QKI 蛋白的表达进行检测。
1.2.6 Western blot 检测 将收集到的细胞样品按照RIPA 裂解液操作说明处理得到蛋白样品,使用BCA蛋白浓度测定试剂盒测定总蛋白浓度。上样等质量的变性后的蛋白样本进行SDS-PAGE 凝胶电泳,电泳结束后将蛋白转印至PVDF 膜上,使用10%的脱脂奶粉封闭2 h。封闭完成后,TBST 洗膜3 次/5 min,分别用兔抗QKI 抗体(1∶1 000 稀释)和兔抗GAPDH 抗体(1∶5 000 稀释)4℃摇床孵育过夜,TBST 洗膜3 次/5 min,再加入HRP 标记的山羊抗兔二抗(1∶5 000 稀释)中,室温摇床孵育2 h,TBST 洗膜3 次/5 min,之后将膜放入ECL 化学发光液中作用2 min,凝胶成像系统曝光并分析结果。
1.2.7 免疫荧光染色观察QKI 蛋白的表达分布 使用4%多聚甲醛固定Quaking 基因敲除NIH3T3 细胞样品及对照组细胞样品,PBS 清洗后再用0.1%Trition X-100 对细胞进行通透处理;处理后的细胞加入1% BSA 进行封闭,随后使用QKI 抗体(1∶100稀释)进行过夜孵育,次日PBS 清洗后再加入绿色荧光标记的Donkey anti-Rabbit IgG(H+L)Highly Cross-Adsorbed Secondary Antibody(Alexa Fluor 488,1∶1 000 稀释)孵育。孵育结束后,使用DAPI 对细胞进行染核,PBS 清洗后封片并在荧光显微镜下观察并拍照。
做好雨污分流,有利于减少污水产生量,降低运营成本,有利于降低垃圾堆体含水率,减少臭气产生量,提高堆体稳定性,是实现生活垃圾卫生填埋的关键所在。
1.2.8 细胞增殖试验 将生长状态良好的Quaking基因敲除NIH3T3 细胞及对照组细胞分别以2 × 103个/孔的密度接种于96 孔板,分别于第1、2、3、4、5、6、7 天加入10 μL CCK8 试剂,继续在37℃培养箱培养2 h 后使用酶标仪检测450 nm 处的吸光度值,每组6 个复孔,统计结果并绘制细胞生长曲线。使用GraphPad Prism 8.0 软件进行统计学分析,数据以平均值±标准误表示,两组间样本采用双因素方差分析,*P < 0.05 表示差异有统计学意义,**P < 0.01表示差异显著,***P < 0.001 表示差异极显著。
2 结果
2.1 Quaking基因sgRNA靶点序列的设计
将NCBI 数据库中获取的小鼠Quaking 基因的基因组序列导入SnapGene 软件绘制基因序列图谱,利用CRISPR 在线设计网站分别在Quaking 基因的第1、2 外显子区域设计sgRNA。选择第1、2 外显子分别作为两个敲除位点,对应sgRNA2 和sgRNA1(图1)。
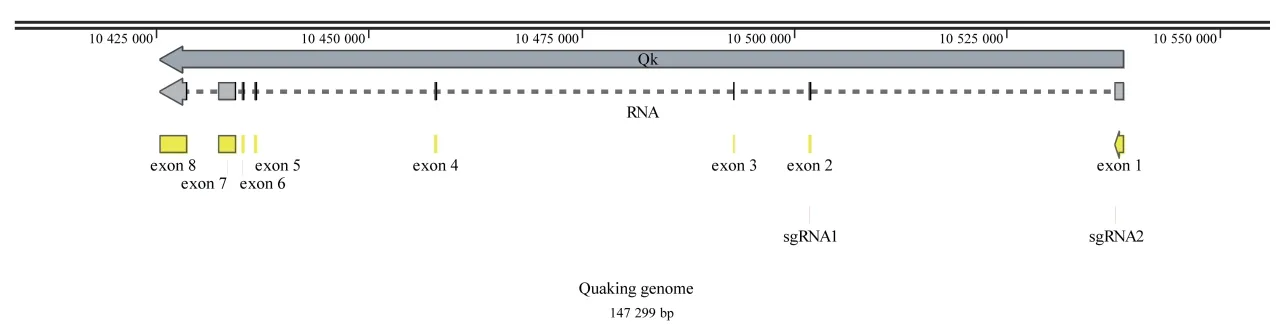
图1 小鼠Quaking 基因的sgRNA 设计图Fig.1 sgRNA design of Quaking gene in mice
2.2 重组质粒LentiCRISPRv2-sgRNA的构建与鉴定
使用限制性核酸内切酶Esp3I 酶切线性化LentiCRISPRv2 空载体,由于LentiCRISPRv2 载体上存在2 个Esp3I 酶切位点,故Esp3I 酶切后可获得两个条带。如图2-A 所示,Esp3I 酶切后成功获得与预期大小相符的两个条带(12 984 bp 和1 899 bp)。随后,利用T4 DNA Ligase 将两对sgRNA 退火产物分别与酶切线性化的LentiCRISPRv2 空载体连接构成LentiCRISPRv2-sgRNA1 和LentiCRISPRv2-sgRNA2重组质粒。将两个重组质粒进行测序,测序结果显示靶向Quaking 基因的sgRNA1 和sgRNA2 已经成功插入到LentiCRISPRv2 载体(图2-B),上述结果表明成功构建重组质粒LentiCRISPRv2-sgRNA1 和LentiCRISPRv2-sgRNA2。
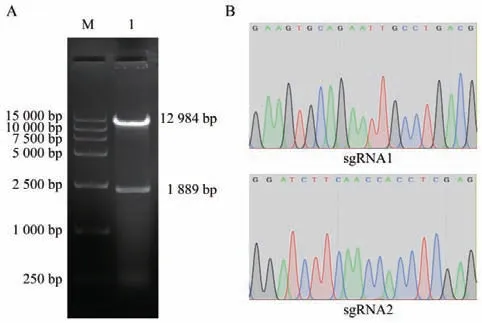
图2 重组质粒的构建与鉴定结果Fig.2 Construction and identification of recombinant plasmids
2.3 筛选QKI蛋白敲除效率高的重组质粒
复苏并传代培养HEK293T 细胞,将重组质粒LentiCRISPRv2-sgRNA1 和LentiCRISPRv2-sgRNA2分别与pcDNA3.1-Quaking 质粒共转染至HEK293T细胞中,并以未转染的空白细胞作为对照组。转染后48 h 收取细胞蛋白样品,用于Western blot检测,以筛选敲除效率高的重组质粒。如图3 所示,第4 泳道的LentiCRISPRv2-sgRNA1 的敲除效率高于第5 泳道的LentiCRISPRv2-sgRNA2,故选择LentiCRISPRv2-sgRNA1 重组质粒用于后续试验。
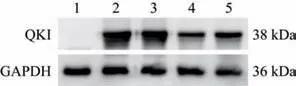
图3 Western blot 筛选QKI 蛋白敲除效率高的重组质粒Fig.3 Screening of recombinant plasmids with high QKI protein knockout efficiency via Western blot
2.4 NIH3T3细胞嘌呤霉素最低致死浓度筛选
为了获得慢病毒转导后经药物筛选的阳性细胞,设置不同梯度浓度的嘌呤霉素,将它们分别加入状态良好的NIH3T3 细胞中,观察细胞的存活状态并进行记录,最终获得48 h 内使NIH3T3 细胞全部死亡的最低嘌呤浓度为1.5 μg/mL,后续使用该浓度进行单细胞克隆的筛选。
2.5 Quaking基因敲除的NIH3T3细胞株的建立
将敲除效率高的LentiCRISPRv2-sgRNA1 重组质粒与辅助包装质粒psPAX2、pMD2.G 共转染至HEK293T 细胞中进行慢病毒包装。使用包装好的慢病毒毒液感染NIH3T3 细胞,感染48 h 后加入浓度为1.5 μg/mL 的嘌呤霉素以筛选阳性细胞。将筛选后的阳性单克隆细胞扩大培养后,提取单克隆细胞的基因组DNA 并PCR 扩增靶位点附近序列。测序结果如图4 所示,与野生型细胞株相比,单克隆细胞株在sgRNA 序列中缺失了7 个碱基造成移码突变,表明成功建立Quaking 基因敲除的NIH3T3 细胞株。在普通光学显微镜下观察细胞形态发现,Quaking 基因敲除的NIH3T3 细胞与正常对照组细胞形态无差异(图5)。

图4 Quaking 敲除细胞株与野生型细胞株测序结果比对示意图Fig.4 Schematic diagram of comparison between sequencing results of Quaking knockout cell lines and wildtype cell lines
图5 NIH3T3 细胞形态图Fig.5 Morphology of NIH3T3 cells
2.6 Western blot检测Quaking基因敲除细胞株中QKI蛋白的表达情况
使用Western blot 检测小鼠Quaking 基因敲除细胞株中QKI 蛋白的表达,结果如图6 所示,与对照组相比,Quaking 基因敲除细胞株中未检测到QKI蛋白的表达,进一步从蛋白水平证明Quaking 基因敲除的NIH3T3 细胞株构建成功。
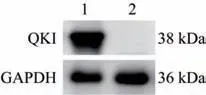
图6 Western blot 检测QKI 蛋白表达Fig.6 Detection of QKI protein expression via Western blot
2.7 免疫荧光染色观察Quaking基因敲除细胞株中QKI蛋白的表达分布
利用免疫荧光染色技术观察Quaking 基因敲除NIH3T3 细胞株中QKI 蛋白的表达分布,结果如图7所示,对照组NIH3T3 细胞中QKI 蛋白主要表达在细胞核中,而Quaking 敲除的NIH3T3 细胞中未见绿色荧光,即无QKI 蛋白的表达,这些结果进一步证明Quaking 基因敲除的NIH3T3 细胞株构建成功。
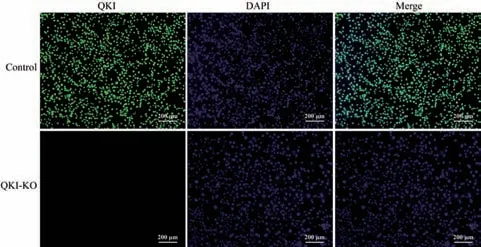
图7 免疫荧光染色检测QKI 蛋白表达Fig.7 QKI protein expression detected by immunofluorescence staining
2.8 Quaking敲除抑制NIH3T3细胞的增殖能力
为了探讨QKI 蛋白的敲除对NIH3T3 细胞增殖的影响,采用CCK8 对生长状态良好的Quaking 基因敲除的NIH3T3 细胞及对照组细胞的增殖活力进行测定。结果如图8 所示,Quaking 基因敲除细胞株的增殖活力较对照组细胞极显著降低(P < 0.001),但在第7 天时两组细胞的增殖活力趋于一致。
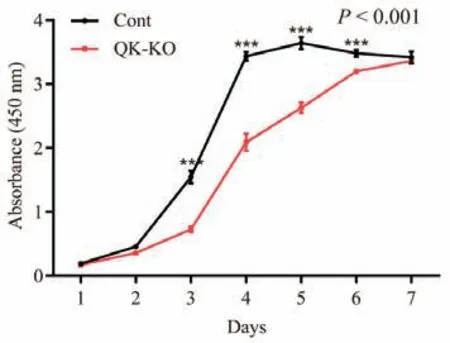
图8 Quaking 基因敲除对NIH3T3 细胞增殖能力的影响Fig.8 Effects of Quaking gene knockout on the proliferation ability of NIH3T3 cells
3 讨论
CRISPR/Cas9 基因编辑技术作为一种可在全基因组范围进行高通量筛选的新型基因编辑技术,具有操作简单、成本低、高效性和低脱靶率的优点,可实现对基因组的精准修饰[16]。自2013 年CRISPR/Cas9 基因编辑技术在人类细胞中得到验证以来[17-19],sgRNA 引导的CRISPR/Cas9 系统已应用于多种细胞系和生物体的DNA 和RNA 操作中,并在疾病免疫机理、药物靶点筛选和动物遗传育种等领域获得广泛应用[17,20-21]。在可用于精确基因治疗的工具中,CRISPR/Cas9 系统与锌指核酸酶(zincfinger nucleases,ZFNs)或转录激活因子样效应物核酸酶(transcription activator-like effector nucleases,TALENs)等传统的基因编辑工具相比更简单、更省时,因为该系统只需要一个Cas9 核酸内切酶和一个短的sgRNA[22]。慢病毒是一种逆转录病毒,它可将大量的病毒互补DNA 整合到宿主细胞的基因组中,并且可以有效地感染分裂期及非分裂期细胞,是最有效的基因传递方法之一[23-24]。因此,本研究选择同时包含有CRISPR/Cas9 系统和慢病毒载体筛选系统的LentiCRISPRv2 质粒进行敲除细胞株的构建。
尽管CRISPR/Cas9 技术日益成熟,但其安全性和效率仍然是需要重点考虑的问题[25]。sgRNA 往往具有相对较高的错配耐受性,Cas9 通常会切割与靶基因序列相似的脱靶位点,这就造成了CRISPR/Cas9 系统的实际应用一直受到脱靶效应的影响[26-27]。为了最大程度降低脱靶效应的影响,同时提高靶基因sgRNA 的特异性,本研究利用CRISPR在线设计网站(http://crispor.tefor.net/)设计获得了两个评分最优的分别靶向Quaking 基因第1、2外显子区域的sgRNA。为进一步筛选得到敲除效率更高的sgRNA,本研究首先在更容易转染的HEK293T 细胞系中进行了预实验。在HEK293T 细胞中外源性过表达小鼠QKI 蛋白的同时分别共转染LentiCRISPRv2-sgRNA1 和LentiCRISPRv2-sgRNA2重组质粒,Western blot 检测发现LentiCRISPRv2-sgRNA1 组较LentiCRISPRv2-sgRNA2 组QKI 蛋白的敲除效率更高。随后使用LentiCRISPRv2-sgRNA1 重组质粒进行慢病毒包装,感染NIH3T3 细胞后,经嘌呤霉素筛选获得阳性单克隆细胞,对扩大培养的细胞进行Western blot、免疫荧光染色和测序鉴定,结果均证明成功构建了小鼠Quaking 基因敲除NIH3T3 细胞模型。
RNA 结合蛋白QKI 在心脏、肺脏、脑、视网膜、前列腺、睾丸等多种器官中广泛表达,在调控RNA形成与信号转导等过程中发挥重要功能[4]。QKI 与细胞增殖密切相关,其能有效抑制前列腺癌和肺癌细胞的增殖[5,28],过表达QKI 还可以抑制乳腺癌和卵巢癌细胞的增殖[6-7]。另有研究报道QKI 可以促进血管内皮细胞、睾丸支持细胞的增殖[12,29],过表达QKI 也能促进成肌细胞的增殖[30]。本研究对Quaking 敲除的NIH3T3 细胞株及对照组细胞进行了CCK8 实验,结果显示Quaking 基因的敲除抑制了NIH3T3 细胞的增殖能力,即QKI 的存在可以促进NIH3T3 细胞的增殖,这与前人研究的正常细胞中所观察到现象相吻合,但QKI 调控NIH3T3 细胞增殖的具体机制仍有待于进一步深入研究。除此之外,本研究中所获得的敲除细胞株还可用于研究Quaking 基因的调控网络。另外,可通过比较正常细胞和Quaking 基因敲除细胞的差异,来识别潜在的疾病相关基因,从而有助于研究疾病的发病机制和治疗方法。Quaking 基因敲除细胞株还可用于肿瘤研究,了解Quaking 基因敲除对肿瘤发生和发展的影响,从而为癌症治疗提供线索。总之,Quaking 基因敲除NIH3T3 细胞株的构建,为进一步阐明Quaking对NIH3T3 细胞增殖的调控机制及其他功能研究提供了体外细胞模型。
4 结论
本研究利用CRISPR/Cas9 基因编辑技术成功构建了Quaking 基因敲除的小鼠胚胎成纤维细胞株,并通过细胞增殖试验表明,Quaking 基因敲除显著抑制了NIH3T3 细胞的增殖能力。
猜你喜欢
新民周刊(2022年27期)2022-08-01
传染病信息(2021年6期)2021-02-12
食品科学(2018年10期)2018-05-23
西南医科大学学报(2015年1期)2015-08-22
医学研究杂志(2015年11期)2015-06-10
中国当代医药(2015年16期)2015-03-01
中国当代医药(2015年9期)2015-03-01
中国医药导报(2015年27期)2015-02-28
生物医学工程学进展(2015年1期)2015-02-28
化学工业与工程(2015年1期)2015-02-10
