
植物生长发育动态QTL 解析研究进展
2024-03-12 14:31付威韦素云陈赢男
生物技术通报 2024年2期
付威 韦素云 陈赢男
(林木遗传育种全国重点实验室 现代南方林业协同创新中心 林木遗传与生物技术教育部重点实验室 江苏省林木遗传和高效培育重点实验室 南京林业大学,南京 210037)
生物体的性状主要分为质量性状和数量性状。其中,质量性状是一种不连续性变异,能够被明确分组,通常由一对或几对等位基因控制,不易受环境影响而发生变化[1],植物许多重要性状如性别[2]、育性[3]、色泽[4]等均表现出质量性状遗传的特点。不同于质量性状,数量性状的遗传效应是由一个或几个主效基因和大量微效基因共同控制的,表现出连续的变化,单个控制基因只有微小的控制效应,这种现象被称为多基因效应,数量性状控制基因在染色体上的位置则被称为数量性状基因座[5](quantitative trait locus,QTL)。植物的大部分性状都属于数量性状,如株高[6]、品质[7]、产量[8]、抗病性[9]等,均表现出连续变异的遗传特性。然而,数量性状的形成不仅是多基因相互作用与调控的结果,还受到时间与环境因素的影响,使生物体呈现具有时空规律的生长发育变化,如树木的高生长与粗生长[10]、果实产量的变化[11]、籽粒营养物质的积累[12]等生物进程,这种在生物体生长发育过程中随时空变化而变化的性状就被称为动态性状,也称纵向性状或函数值性状,是基因有选择的时序性表达、细胞群内部和之间互作、基因与环境互作以及表观遗传相互作用的动态表现结果[13]。
自1865 年孟德尔遗传规律发现以来,基因定位及其功能验证一直是分子遗传学研究领域的重点与热点,尽管反向遗传学能够有针对性地对基因功能进行研究,但通过生物体的表型差异定位目标基因并研究其功能的正向遗传学策略仍是开展基因定位与功能研究的最有效方式[14]。目前,数量性状基因定位根据其原理主要分为基于连锁规律的连锁分析(linkage analysis)和基于连锁不平衡定律的关联分析(association analysis)。QTL 定位实质上是利用连锁遗传规律对家系内子代的目标性状与分子标记的连锁关系进行分析,从而明确目标性状QTL 在染色体上的位置。通过QTL 作图,可以建立起已知分子标记与未知QTL 之间的连锁关系,从而在遗传图谱上定位连锁群(linkage group)与功能基因(图1)。1988 年,Paterson 等[15]首次在番茄中应用RFLP 遗传图谱定位到了控制番茄果实质量、可溶性固形物以及果实pH 的QTL,从此数量性状的定位进入分子标记的时代,鉴定了许多复杂性状QTL[16-18]。常规QTL 方法,即非条件QTL(unconditional QTL)采取“静态定位”(static mapping)的策略[19],对植株性状的发育终点进行分析,定位到的目标性状连锁群只能反映植株某一特定生长阶段基因表达的累积效应,是性状表达的最终表现状态[20]。然而,对数量性状而言,性状的表达是连续的,每个时期的表型值都是多个QTL 动态表达的结果,从而呈现连续变异的遗传效应,常规QTL 方法定位的连锁群与候选基因忽略了只在某一时期特异表达的QTL,既不能反映目标性状QTL 在植株生长发育过程中的真实表达情况,也无法分析多个相关性状间的遗传关系,具有很大的局限性[21]。因此,在常规QTL 的基础上,吴为人等[20]和Zhu 等[22]均提出了QTL 的动态定位(dynamic mapping)策略,即条件QTL(conditional QTL),通过检测多个测量时期内数量性状的净增长量,分析特定发育阶段QTL 的净效应,从而有效利用生物体生长发育过程中的遗传信息,提高定位准确性与灵敏性,实现动态QTL 的精细定位。邬荣领团队提出的功能作图方法将个体发育过程中影响发育的QTL 效应看作时间函数,强调随时空变化的性状发育规律,从而定位复杂性状的相关基因,解析基因受时间与环境变化而发生的变化,实现动态QTL 定位及对发育性状的遗传调控解析[23]。
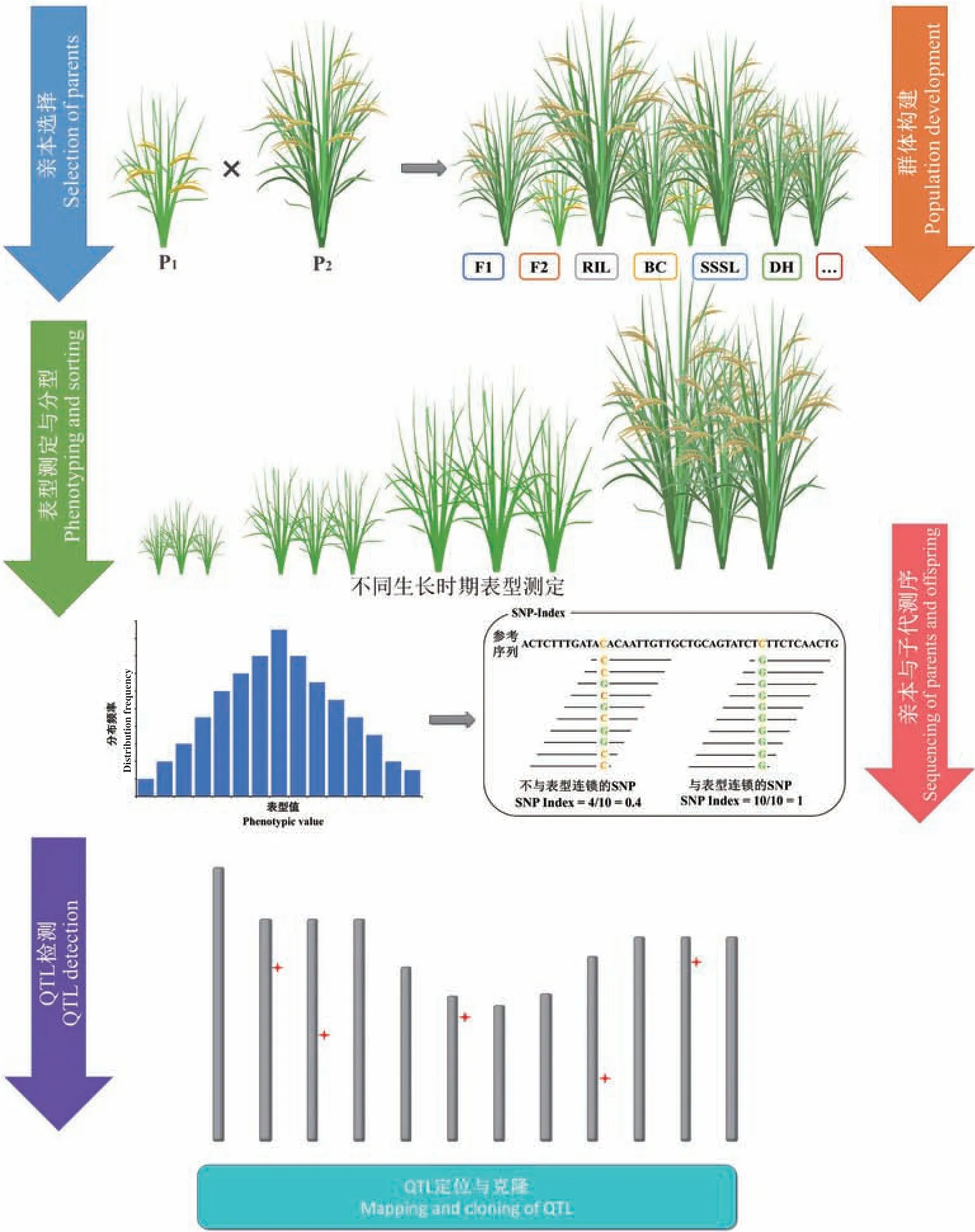
图1 动态QTL 分析流程图Fig.1 Flow chart of dynamic QTL analysis
1 动态QTL 研究模型
植物性状是各分量综合发育的结果,模型则是对特定系统及特征的简洁描述与概括[5],通过建立生物体动态发育性状的遗传模型,可以研究遗传和非遗传因素及其相互作用对表型变异的影响、个体发育的变化规律以及选择响应的大小、方向及维度等[24]。目前,已经建成的发育遗传模型主要有3 种:直接效应模型、主-多基因混合遗传模型和生长轨迹模型[13]。直接效应模型属于经典的孟德尔遗传模型,包括适用于单一基因控制简单性状的单基因模型和受少数几个主基因控制性状的寡基因模型。直接效应模型认为个体的遗传信息完全遗传自亲本的基因组,所有遗传分量仅限于直接遗传分量(如基因的显性效应和加性效应),并且个体发育不受环境因子和渐成因子的影响,性别之间完全相关等[25]。通过杂交子代的性状分离比例,可以剖析性状控制基因的遗传效应大小,对个体发育的最终表现做出预测和评价。但生物体发育是一个受多因素影响的复杂生物进程,不仅受遗传基因控制,还受环境因子、母体效应、渐成效应及表观遗传效应的影响,是一个互作、渐成的过程[26-27]。利用直接效应模型研究动态发育的数量性状,不足以体现数量性状的动态发育,也不足以解释各遗传分量在性状发展过程中的遗传贡献及遗传与非遗传因素间的互作和变化规律[28]。因此,直接效应模型不适于解释多基因控制的动态发育数量性状[13]。
对动态发育性状而言,其遗传模型必须同时反映其遗传和发育本质,并提供影响遗传变异及发育进程的相关因素,体现发育性状互作、连续、可遗传、渐成的特点[28]。早期对动态性状的遗传研究集中于某个时间节点或粗略划分的几个时间段,且只考虑最终性状的表现值,建立的模型也只是对静态现象的简单描述。但生物体在不同的发育阶段,基因表达不同,遗传机理会发生变化,使数量性状呈现出具有时空规律的动态表现[29]。此外,控制数量性状的基因众多,不同基因座对表现型的贡献不同,基因之间又存在相互作用,而且基因表达易受环境的影响[21]。因此,仅研究最终性状的遗传模型不足以解释复杂数量性状建成过程中的遗传作用规律,需要选用能够反映数量性状动态发育过程的遗传模型。
1.1 主-多基因混合遗传模型
微效遗传假说认为数量性状受多基因控制,单个基因只有很小的遗传效应。但很多试验数据表明,控制数量性状的基因遗传效应并不相等,其中遗传效应较大的为主效基因,效应较小则为微效基因,构成了控制数量性状的主-多基因遗传系统[24]。通过观察分离世代的性状分布特征可以判断是否存在主基因,即无主基因的多基因体系呈现单峰对称的正态分布,而存在主基因时则会呈现多峰或偏离正态分布的单峰分离[1]。例如小麦(Triticum aestivum)的株高和产量[30]、菊花(Chrysanthemum morifolium)的营养性状[31]、水稻(Oryza sativa)的株高[32]、油菜(Brassica rapa var.oleifera)的分支角度[33]、苦瓜(Momordica charantia)的抗病性[34]、陆地棉(Gossypium hirsutum)的果枝夹角[35]等,主基因使性状分布频率成峰,微效基因和随机环境效应使其连续,盖均镒[36]将其概括为主-多基因遗传体系。对于主基因的鉴别,主要通过复合分离分析方法,参数估计采用极大似然法,计算方法采用EM 算法;主基因的检测采用似然比测验,已广泛应用于人类和动物遗传病的研究[37]。
在植物中,Elkind 等[38]提出了一个包含主基因和多基因的遗传模型,使后代的基因性质可明确分类。然而,这种分析方法只包括了1 对主基因和多基因的遗传模型,并未考虑2 对甚至多对主基因和连锁及上位效应。因此,盖钧镒[24]建立了“植物数量性状遗传体系主基因-多基因混合遗传模型分离分析法”,其基本思路与环节包括:(1)分离世代的理论分布与基本假设;(2)7 类遗传模型;(3)单世代或多世代分析;(4)建立似然函数;(5)估计分布参数;(6)选择最适模型;(7)估计遗传参数;(8)判别个体基因型;(9)图形分析。通过“植物数量性状遗传体系主基因-多基因混合遗传模型分离分析法”能够得出主、多基因的相对重要性、效应大小、基因作用方式等,对于植物数量性状遗传模型发展具有十分重要的理论和现实意义。汪文祥等[33]在油菜BCP1、BCP2 和F2群体中对其分支角度进行了遗传分析,鉴定到顶枝角主基因的遗传贡献率分别为34.08%、1.40%、14.99%,存在明显的主效基因;谷子[39](Setaria italica var.germanica)的株高、穗长与穗粒重等均符合主-多基因混合遗传模型,其中穗长的主基因遗传贡献率为46.40%,多基因遗传贡献率为46.91%,而穗粒重的主基因遗传贡献率高达81.10%。主-多基因混合遗传模型包含了复杂的统计和计算,对实验设计、误差检验、群体大小等方面都有严格的要求,模型难以简化,否则会对鉴别精度产生影响,因此一般应用于遗传效应明显且数量较少的主基因鉴定[24]。
1.2 生长轨迹模型
生物体很多性状的表现都随着时间变化而呈现一定的规律和连续性,对同一性状不同时间点的表型值进行测量,会得到表型值与时间之间的函数,从而生成一条光滑的曲线,这条曲线被称为“生长曲线”,可以用来描述和解释生长发育的规律,而复杂性状的各分量则有各自的发育轨迹[23,40-41]。通过将重复测量的表型数据拟合生长曲线,基于降维的多元分析理论,可以将复杂的动态性状分解为各分量的发育动态,从而对少量生长参数进行遗传分析[42]。Kirkpatrick 等[43]在对小鼠早期生长分析的试验中提出了一种估算遗传和选择参数的连续定量遗传模型,Atchley 等[13]在此基础上将不同的分量综合成复杂数量性状的生长轨迹模型:dxi(t)/dt=Fi[x(t)]xi(t),其中dxi(t)/dt 为细胞发育群体在时刻表型值的向量表示,xi(t)表示t 时刻细胞群体包含的细胞总数,各分量的生长轨迹Fi[x(t)]则通过常量ai、系数bij及初始条件表示为通过该模型,不仅可以获取生长模型所必需的发育信息,还能够对生长发育做出遗传解释。发育遗传学中,常用的生长模型有Gompertz 方程、Logistic 方程以及 Richards 方程等[44],其中Logistic 模型又称“S 型曲线”或“生长曲线”,常用于描述生物体的增长特征:g(t)=a/(1+be-rt),其中g 表示遗传值,t 为时间,当t →∞时,a 是g 的渐近值,r 是相对生长率,当t →0 时,a/(1+b)共同表示g 的初始值,(a,b,r)3 个参数的组合决定了生长曲线的整体形状,后续还可以转换成加性效应和显性效应。Wu 等[40]基于logistic 模型提出了一种QTL 定位的“两步法”:首先用动态性状的表型值拟合生长曲线,然后对估算的生长参数采用复合区间作图进行动态性状的QTL定位。邬荣领团队对两步法作了改进[23,41,45],提出了基于logistic 生长模型[42]的“一步法”,精确定位生物体发育过程中的动态性状。生长轨迹模型有效地综合了数量性状各分量的发育动态,从而获得生物体生长发育过程中的重要发育信息。
2 动态性状的分析方法
2.1 条件分析法
1995 年,朱军团队提出并发展了基于复合区间作图的混合线性模型(composite interval mapping based mixed linear model,MCIM)及针对发育性状的条件QTL 分析法,从而将QTL 的各项遗传效应(显性效应、加性效应、上位性效应等)、随机环境效应、基因与环境互作效应等纳入数量性状的统计模型,将遗传效应估计与QTL 定位相结合,从而实现QTL 的精确定位及对动态发育性状的遗传分析[46-49]。条件分析法对(t-1)至t 时刻的遗传效应净增量进行分析,对于给定(t-1)时刻的表型值,t 时刻的表型值则为条件随机变量,模型表示为,yhjk(t|t-1)=μ(t|t-1)+Gj(t|t-1)+Eh(t|t-1)+GEhj(t|t-1)+Ehjk(t|t-1),通过条件随机效应预测可获得条件遗传效应Gj(t|t-1)及条件互作效应GEhj(t|t-1)的无偏预测值后,利用复合区间作图法分析yhjk(t|t-1)=μ(t|t-1)+Gj(t|t-1)特定发育阶段(t-1)→t 时刻的净遗传主效应,再估计yhj(GE)(t|t-1)=μ(t|t-1)+Eh(t|t-1)+GEhj(t|t-1)的预测值,分析(t-1)→t时刻QTL 或基因与环境互作效应。条件分析法已在小麦[6,50-53]、水稻[19,54]、玉米(Zea mays)[55-56]和油菜[12,16,57]的动态发育性状研究中广泛应用。
2.2 功能作图法
随着分子标记技术、高通量测序技术以及生物信息学方法的发展与完善,邬荣领团队将统计学、分子遗传学与生物生长发育机制相结合,提出了复杂动态性状QTL 的定量分析方法[23],即功能作图(functional mapping),将反映发育生物学原理的模型与统计方法引入基因定位,强调性状建成过程中基因型与环境的互作效应,通过EM 算法实现对QTL位置和遗传效应的最大似然估计,使得复杂数量性状的遗传机制解析成为可能[58]。以回交群体为例,首先对多时间点测量的动态性状拟合生长曲线,使群体基因型值分布满足多元正态分布:
gj为经logistic 模型优化的基因型值:
∑为不同时间点测量表型值的残差方差及协方差矩阵,通过自回归预测法(autoregression,AR)对其重复测量误差建模并假设矩阵Σ 方差稳定得出:
直至收敛,此时的收敛值就是Ω 的最大似然估计。与传统的QTL 定位相比,功能作图在以下方面有着显著优势[21]:(1)考虑了表型性状的动态建成过程,其结果更贴近生物学事实;(2)将复杂发育性状QTL 按作用时长、起始时间划分为长效QTL(long QTL)、短效QTL(short QTL)以及反向QTL(inverse QTL),其中短效QTL 分为早期QTL(early QTL)与晚期QTL(late QTL);(3)功能作图对个体表型值在不同时间节点的多次重复测量不仅提升了QTL 定位的精度与准度,通过拟合生长曲线分析大量遗传变量,增强了模型的普适性和简洁性。
3 动态QTL 研究进展
在动态QTL 定位方法出现之前,对发育性状的QTL 定位主要集中在某个特定生长阶段,无法反映数量性状的动态变化过程。而动态QTL 定位可以估算QTL 在不同生长阶段的实际效应,了解基因在不同发育时期的动态表达[59]。目前,国内外许多学者对植物复杂发育性状进行了大量动态QTL 定位研究(附表1)。Wang 等[57]以3 个地点不同时期油菜的DH 群体(共348 个株系)为研究材料,对其不同发育时期的株高发育动态进行了非条件及条件QTL 分析。结果表明,在油菜成熟期共鉴定到31 个非条件QTL,而在油菜生长的6 个时间段,共鉴定到50 个条件QTL,其中只有5 个非条件QTL(ucPH.A2-2、ucPH.A3-2、ucPH.C5-1、ucPH.C6-2、ucPH.C6-3)和1 个条件QTL(cPH.A2-3)在油菜生长的整个发育时期都有表达,是控制油菜株高的长效QTL,并证实了数量性状是QTL 选择性表达的结果。Wu 等[60]在葡萄(Vitis vinifera)果形的动态QTL 分析中同样鉴定到1 个长效QTL(qShI-5-1),在3 次试验中均有表达,解释了葡萄果实形状16.4%-19.8%的表型变异。Miedaner 等[61]对杂交黑麦(Secale cereale)的株高及生物量进行了条件QTL 研究,定位到株高的18 个条件QTL 和生物量的3 个条件QTL,但没有一个QTL 在测定的3 个发育时期内都有表达,体现了发育数量性状的阶段表达特性。An 等[10]对橡胶树(Hevea brasiliensis)‘CATAS 8-79’与‘MT/C/11 9/67’杂交产生的F1代个体(206 株)的橡胶产量和胸径生长的发育动态进行了非条件及条件QTL 分析,鉴定到7 个控制橡胶产量和6 个控制胸径生长的加性条件QTL,其中QTL qLD-13-1 既在S3 阶段被传统QTL 分析检测到,同时在S3|S2 阶段通过条件映射方法也被鉴定,说明qLD-13-1 对S3 阶段前期的乳胶产量影响不大,而对S3 阶段的乳胶产量有显著影响,在S3|S2 阶段则产生了条件净效应。
Zhang 等[62]以花生(Arachis hypogaea)品种YZ9102 与wt09-0023 杂交构建的包含521 个株系的RIL 群体为材料,对花生新鲜种子发芽过程进行了动态QTL 定位,共检测到24 个条件QTL,解释了亲本0.82%-15.83%的表型变异,其中一个QTL qFSGA04 在3 种发芽环境的整个发芽阶段均能表达,是控制花生新鲜种子发芽的主效与长效QTL,其他则为短效QTL。在玉米中,Li 等[63]对玉米穗丝发生后细胞壁组分和饲料消化率的变化进行了动态QTL 分析,在五个阶段共检测出72 个非条件QTL和26 个条件QTL,其中6 个细胞壁组分变化的主效条件QTL 解释了超过10%的表型变异。Zhang 等[64]对小麦品种Huapei 3 与Yumai 57 杂交产生的DH 群体(168 株)的穗数、每穗粒数和千粒重进行条件QTL 分析,共检测到59 个条件QTL,并得出不同QTL 间相互作用的结论。Bu 等[65]在对毛豆(Glycine max)种子发育的QTL 定位中发现,共有12 个非条件QTL 与8 个条件QTL 在毛豆种子发育过程中显著影响种子硬度的变化,其中鉴定到的非条件QTL 与条件QTL 大多不同,共同的QTL 比例较少,非条件QTL 与条件QTL 的数目均呈现先增加后减少的趋势,说明大多数QTL 的表达是按照一定时空规律阶段性表达的。Su 等[66]在对菊花(C.morifolium)耐涝性的遗传机制解析中发现,控制菊花耐涝的非条件QTL 与条件QTL 个数分别为37 个与51 个,并鉴定到大量的上位效应QTL。李海洋[67]连续两年对烟草(Nicotiana tabacum)RIL 群体的青枯病抗性作了动态QTL 定位,共检测到9 个非条件QTL 与13个条件QTL,在2016 年定位到3 个非条件QTL 与8 个条件QTL,2017 年定位到7 个非条件QTL(含1 个重复)与5 个条件QTL,分别于2016 年和2017年检测到1 个长效QTL 在多个调查期持续表达,但仅有1 个QTL(qBWR2-1)能在不同调查期被重复检测到,表明多数QTL 的表达具有时序性。
Xie 等[68]以水稻的重组自交系群体为研究材料鉴定了低温(15℃)和最适温度(25℃)条件下显著影响水稻种子萌发和幼苗形态建成的动态QTL,结果表明,显著影响种子活力的条件QTL 与非条件QTL 不尽相同,共检测到了5 个非条件QTL(qSV-1、qSV-5a、qSV-5c、qSV-8、qSV-11)与3 个条件QTL(qSV-5b、qSV-6a、qSV-6b),其中qSV-1、qSV-5b、qSV-6a、qSV-6b 仅影响水稻幼苗形态建成,qSV-5a、qSV-5c、qSV-8 仅对种子萌发起促进作用,而非条件QTL qSV-11 在种子萌发及幼苗形态建成中均能发挥作用;3 个条件QTL 仅在低温条件下幼苗形态建成的第3 阶段显著表达,被鉴定为水稻幼苗形态建成的低温特异性QTL 及短效QTL,该研究为进一步了解水稻种子萌发及幼苗形态建成奠定了基础。Bu等[69]对水稻HJX74 的5 个单片段替换系(singlesegment substitution lines,SSSLs)39 种基因型的叶片数量进行了条件QTL 研究,在9 个发育阶段中共检测到5 个具有显著加性效应的条件QTL,均对叶片数量增加起促进作用,并且在其中3 个时间段(t0-t2、t4-t6、t7-t9),5 个条件QTL 的表达更加活跃。Zheng 等[70]对水稻品种Asominori 与IR24 杂交产生的RIL 群体71 个子代进行了动态QTL 分析,检测到了籽粒蛋白含量的10 个非条件QTL 和6 个条件QTL,其中仅有1 个条件QTL qPC-9 在两个发育阶段连续表达,其余5 个条件QTL 仅在单个发育时期特异性表达,且条件QTL 的数目随发育时期延长呈现先增后减再增的变化;在对水稻籽粒蛋白指数的QTL 定位研究中发现,检测到的非条件QTL 数目随着发育时期逐渐减少,说明水稻籽粒蛋白合成基因在籽粒发育前期更加活跃;结合非条件QTL 和条件QTL 分析检测到了7 个共同QTL 影响籽粒蛋白指数,共同QTL 的比例较高,并且大部分QTL 在籽粒发育前期而非成熟期被检测,说明多数QTL(基因)的表达是动态的、分阶段的,通过全生长周期的动态QTL 分析比常规QTL 定位检测到的QTL 种类与数目更加全面。
Zhang 等[71]在对杨树杂交种(Populous deltoides×P.canadensis syn.euramericana)扦插生根的遗传定位研究中,鉴定到插穗最大根长和总根数的6 个条件QTL,其中仅有1 个QTL 在生根过程中持续影响根系生长,其余5 个短效QTL 对根系生长的影响随生根时间变化而变化,在插穗生根的不同阶段中具有不同的发育模式。在簸箕柳(Salix suchowensis)生长节律的遗传机制解析[72]中,共鉴定到控制株高的2 个长效QTL 和3 个短效QTL,以及控制地径生长的2 个长效QTL 和4 个短效QTL,在簸箕柳生长节律的动态变化中发挥了重要作用。Zhang 等[73]在对胡杨(P.euphratica)根系与地上部协同变化的遗传定位研究中,首先将幼苗生长数据拟合生长方程,从而估算出茎和主根生长的渐近生长、相对生长速率、拐点时间和线性生长的持续时间,通过单变量定位模型检测到15 个茎伸长和4 个介导主根生长的异时QTL(heterochronic quantitative trait loci,hQTLs),通过双变量定位模型共定位到11 个多效性异时QTL,在胡杨地上部与地下部协同生长中发挥重要作用。在对拟南芥(Arabidopsis thaliana)表型可塑性基因定位的研究中,任翔宇[74]检测到世代内显著影响开花时间、果荚数目和叶片数量表型可塑性的位点58、71 和24 个,并且定位到23 个和31 个分别调控开花时间和果荚数目的母体环境表型可塑性显著位点。结果表明,母代受环境影响,可将表型可塑性以基因修饰方式传递给子代,表明QTL 或基因受母体效应和环境效应影响而呈现发育阶段性表达。
4 展望
基因组测序技术和基因编辑技术的迅速发展使定向设计和精准分子育种成为可能。准确检测、鉴定与克隆QTL(基因)并验证其功能是开展精准育种的重要基础,而QTL 定位是主要手段。随着测序通量的提升、新型分子标记的开发及生物信息学方法的发展,使构建植物饱和遗传图谱、实现重要性状QTL 的精细定位成为可能。通过检测在不同遗传背景和环境条件下稳定表达的QTL,利用与目标性状基因连锁的分子标记来筛选目标性状基因型,可以提高QTL 的跟踪效率,缩短育种周期,提高育种效率。然而,许多复杂的发育数量性状如株高、地径、产量的控制基因在不同发育阶段的表达模式不同,是QTL 按一定的时空规律选择性表达的结果。因此,只有通过条件QTL、功能作图等动态QTL 分析方法,才能鉴定目标性状不同发育时期的主效与长效QTL,从而明确QTL 对目标性状的影响模式,实现分子标记辅助选育(molecular marker-assisted selection,MAS)对目标性状的定向改良。
尽管通过动态QTL 分析能够有效检测生物体目标性状不同发育阶段的数量性状控制位点,但受限于人工作图群体亲本的遗传多样性,亲本遗传背景相似度较高,所含可供挖掘优良基因的丰富程度低,子代分离群体获得的重组位点有限,仅用人工作图群体进行的QTL 研究,定位到的QTL 分辨率低、置信区间较大,包含太多的候选基因,不利于后续数量性状控制基因的验证与克隆,分子标记辅助选育的精准度不够。为了保证动态QTL 定位的可信度,多群体、多环境、多时间及多分子标记的组合试验必不可少。对于非模式物种与木本植物而言,研究材料生长周期长、遗传背景复杂、大群体构建困难等问题,严重制约了QTL(基因)的精细定位,仅以单个作图群体定位的数量性状基因座,QTL 定位区间太大,难以满足基因精细定位与克隆转化实验的要求,在后续试验中,应结合多种作图群体同时开展动态QTL 分析,提高QTL 分辨率。结合QTL定位与全基因组关联分析(genome wide association analysis,GWAS)是进行目标性状QTL 定位的新方法,能够鉴定与目标性状显著关联的新位点,有利于发育数量性状位点的精确定位,以自然群体为研究材料,结合人工作图群体,开展重要数量性状的GWAS 与QTL 共定位分析,利用自然群体丰富的遗传资源与基因位点,提高分子标记与基因的连锁程度,缩小QTL 定位范围,从而实现目标性状QTL(基因)的精确定位。随着基因组测序技术的高速发展,利用基于限制性酶切位点关联 DNA 测序(restrictionsite associated DNA sequencing,RAD-seq)、特异性基因座扩增片段测序技术(specific locus amplified fragment sequencing,SALF-seq)等简化全基因组测序方法,能够快速鉴定高密度SNP 位点,结合传统SSR、AFLP 等DNA 分子标记,能够提高遗传连锁图谱分子标记密度,构建高密度遗传图谱,从而提高QTL 定位的效率。此外,QTL 结合转录组测序、代谢组分析等多组学分析技术,能够挖掘控制目标性状动态发育的关键候选基因。随着Meta 分析、Overview 等分析方法的创新,将不同群体、环境和分子标记下定位到的QTL 整合到同一遗传图谱上,从而挖掘出置信区间小、分辨率高、遗传效应显著的QTL 位点,加快实现QTL 的精细定位。
文章所有附表数据请到本刊官网下载(https://biotech.aiijournal.com/CN/1002-5464/home.shtml)。
猜你喜欢
区域治理(2022年40期)2022-11-27
世界科学技术-中医药现代化(2020年2期)2020-07-25
动漫界·幼教365(小班)(2019年10期)2019-10-28
动漫界·幼教365(大班)(2019年10期)2019-10-28
动漫界·幼教365(中班)(2019年10期)2019-10-28
中华家教(2018年7期)2018-08-01
临床医药文献杂志(电子版)(2017年11期)2017-05-17
文学少年(原创儿童文学)(2016年16期)2016-02-28
中学生物学(2016年8期)2016-01-18
中国当代医药(2015年7期)2015-03-01
