
ldentification of key genes regulating the synthesis of quercetin derivatives in Rosa roxburghii through integrated transcriptomics and metabolomics
2024-03-12 13:31LiyaoSuMinWuTianZhangYanZhongZongmingMaxCheng
Liyao Su,Min Wu,Tian Zhang,Yan Zhong,Zongming (Max) Cheng
State Key Laboratory of Crop Genetics and Germplasm Enhancement, College of Horticulture, Nanjing Agricultural University,Nanjing 210095, China
Abstract Rosa roxburghii fruit is rich in flavonoids,but little is known about their biosynthetic pathways. In this study,we employed transcriptomics and metabolomics to study changes related to the flavonoids at five different stages of R.roxburghii fruit development. Flavonoids and the genes related to their biosynthesis were found to undergo significant changes in abundance across different developmental stages,and numerous quercetin derivatives were identified. We found three gene expression modules that were significantly associated with the abundances of the different flavonoids in R.roxburghii and identified three structural UDP-glycosyltransferase genes directly involved in the synthesis of quercetin derivatives within these modules. In addition,we found that RrBEH4,RrLBD1 and RrPIF8 could significantly increase the expression of downstream quercetin derivative biosynthesis genes. Taken together,these results provide new insights into the metabolism of flavonoids and the accumulation of quercetin derivatives in R.roxburghii.
Keywords: Rosa roxburghii,quercetin derivatives,weighted gene co-expression network analysis,transcription factor,transcriptome,metabolome
1.Introduction
Rosaroxburghii,a deciduous shrub of the Rosaceae family,is widely distributed in the southwestern provinces of China,especially in Guizhou Province(Wanget al.2021;Liet al.2022).Rosa roxburghiifruit has a high ascorbic acid content,which has been the subject of several studies (Wanget al.2021;Liet al.2022). However,R.roxburghiifruit also contains numerous flavonoids,as well as vitamin A,carotene,superoxide dismutase,and a variety of amino acids(Wanget al.2021;Liet al.2022).Rosaroxburghiiis consumed as a juice,dried fruit,wine,and in several other forms (Wanget al.2021). Despite the scale of theR.roxburghiiindustry in Guizhou Province,few studies have investigated the molecular mechanisms which control its fruit quality. Therefore,a better understanding of these mechanisms could enable further development of theR.roxburghiiindustry through improved fruit quality.
Flavonoids are a diverse group of metabolites with a C6-C3-C6 carbon frame,which are ubiquitous in the plant kingdom,mainly taking the form of glycosides or carbon sugar groups. The substrates for the synthesis of flavonoids in plants are monoacyl-CoA from the polyketide pathway and 4-coumaryl-CoA produced by the shikimatephenylpropanoid pathway. Phenylalanine generates cinnamic acid by phenylalanine ammonia-lyase (PAL) (Zhuet al.2015),and cinnamic acid is further carboxylated by cinnamate-4-hydroxylase (C4H) to yieldp-coumaric acid(Wanget al.2020). Thep-coumaric acid is converted to coumaroyl-CoA in the presence of 4-coumarate:coenzyme A ligase (4CL) (Liet al.2014). Coumaroyl-CoA and malonyl-CoA are converted by chalcone synthase(CHS) to produce chalcone (Nakajimaet al.1991),which is the basic backbone of all flavonoids. Subsequently,chalcone undergoes isomerization by chalcone isomerase (CHI) to yield naringenin (Ralstonet al.2005).Naringin is the main metabolic precursor in the plant flavonoid synthesis pathway. Naringin forms structurally diverse flavonoid compounds through modifications such as methylation,carboxylation,glycosylation and isopentenylation (Shenet al.2022).
Flavonoids have diverse biological activities and are strong antioxidants due to their phenolic hydroxyl groups(Havsteen 2002;Heimet al.2002;Lucaet al.2020).The flavonoid quercetin has strong antioxidant activity,which is more than 50 times greater than that of vitamin E and 20 times greater than that of vitamin C (Havsteen 2002;Bootset al.2008;Wanget al.2016). In addition,quercetin has been shown to lower blood lipid levels and to possess anti-tumor,anti-hypertensive,and antiaging activities,with no side effects (Lee and Mitchell 2011;Jeonget al.2012). Despite the important roles of flavonoids inR.roxburghiifruit,little is understood about their dynamics during fruit ripening (Yanget al.2020).
In this study,we analyzed the transcriptome and metabolome ofR.roxburghiifruit during development to better understand the dynamics of its flavonoids.Quercetin derivatives were found to accumulate to high levels in matureR.roxburghiifruit,and we used gene expression and metabolic data to construct the metabolic pathways of these derivatives inR.roxburghii.Additionally,we identified several transcription factors that impact the accumulation of quercetin derivatives. Taken together,these results provide a theoretical basis for the development,utilization,and processing of functional compounds fromR.roxburghiiproducts.
2.Materials and methods
2.1.Plant materials and samples
Rosaroxburghiiseeds were planted at the Baima Experimental Station of Nanjing Agricultural University,Jiangsu Province,China. Samples ofR.roxburghiifruit were collected at 10,40,70,100 and 130 days after anthesis and designated as S1,S2,S3,S4,and S5,respectively. Three biological replicates ofR.roxburghiifruit were collected at each time point. After collection,the samples were rapidly frozen in liquid nitrogen and placed in a–80°C freezer for subsequent use.
2.2.Component extraction and determination
To understand the dynamics of metabolites duringR.roxburghiifruit development,we analyzed each time point using targeted metabolomics (Metware Biotechnology Co.,Ltd.,Wuhan,China). Freeze-dried samples ofR.roxburghiiwere ground,and 100 mg of the powder was added to 1 mL of 70% methanol and stored at 4°C overnight. Subsequently,the supernatant was collected by centrifugation at 10,000 r min–1and analyzed using ultra performance liquid chromatography with electrospray ionization coupled tandem mass spectrometry (UPLC-ESI-MS/MS). The components were considered to be differentially accumulated when they met the absolute Log2FC (fold change)≥1 and a VIP (variable importance in the projection)≥1 criteria. We annotated and enriched all metabolites by the Kyoto Encyclopedia of Genes and Genomes(KEGG) compound database (http://www.kegg.jp/kegg/compound/) and the KEGG pathway database (http://www.kegg.jp/kegg/pathway.html).
2.3.Transcriptome sequencing data analysis
Total RNA was extracted using the RNAprep Pure Plant Plus Kit (Tiangen,Beijing,China),according to the manufacturer’s instructions. mRNA libraries ofR.roxburghiifruit were constructed and 200 bp pairedend reads were sequenced on the Illumina NovaSeq 6000 platform. All sequencing data,including genomic and transcriptomic data,can be found in the China National Center for Bioinformation database (PRJCA014460).After removing low-quality reads with fastp (Chenet al.2018),we used HISAT2 (Kimet al.2015) to map the clean reads to theR.roxburghiireference genome (https://ngdc.cncb.ac.cn/,PRJCA014460). FPKM (fragments per kilobase of transcript per million fragments mapped)values for each gene were calculated with featureCounts,and the expression of all samples was combined into one matrix with Trinity (Grabherret al.2013). Differentially expressed genes were identified using the DEseq2 R package with correctedP-value≤0.05 and absolute fold change>1. Finally,the KEGG enrichment analysis of differentially expressed genes was performed with the clusterProfiler R package.
2.4.Co-expression module construction
A weighted gene co-expression network was constructed using the WGCNA R package (Langfelder and Horvath 2008). Flavonoids that were associated with fruit development were utilized as the phenotypic data to identify the related gene expression modules. Additionally,quercetin derivatives were used as the phenotypic data to identify the genes and transcription factors that regulate quercetin synthesis.
2.5.Gene amplification and vector construction
Total RNA was extracted from theR.roxburghiisamples using a plant total RNA isolation kit (Foregene,Chengdu,China),according to the manufacturer’s instructions,and cDNA was synthesized from the total RNA using the PrimeScript RT reagent kit (TaKaRa,Beijing,China).Genomic DNA was extracted with a plant DNA isolation kit (Foregene,Chengdu,China). Subsequently,gene promoter sequences were amplified using genomic DNA and ligated into the pHIS2.1 and pGreenII 0800-LUC vectors,and the genes amplified using cDNA were ligated into the pGADT7-rec and PJX001 vectors. All primers used for amplification are listed in Appendix A.
2.6.Yeast one-hybrid and dual luciferase experiments
To verify the regulatory relationships between the candidate transcription factors and UDP-glycosyltransferase,we transformed the aforementioned vectors together into Y187 yeast receptor cells for yeast onehybrid assays. The transformed yeast cells were cultured in SD–Leu–Trp media (Coolaber,Beijing,China) for 3 days at 30°C. Individual colonies were selected and suspended in double-distilled water,and the concentrations of all colonies were adjusted to OD600=0.05. Finally,the yeast suspensions were transferred to SD–Leu–Trp–His media (Coolaber,Beijing,China) with different concentrations of 3-AT medium and incubated for 4 days.
To verify the results of the yeast one-hybrid experiment,we analyzed the same combinations using dual luciferase assays (YEASEN,Shanghai,China). Candidate transcription factors in PJX001 and the promoters in pGreenII-0800-LUC were transformed intoAgrobacterium tumefaciensstrain GV3101,followed by a 9:1 ratio injection into tobacco leaves. The relative activities of firefly luciferin andRenillaluciferase in tobacco leaves were detected after 3 days.
3.Results
3.1.Metabolomic and transcriptomic profiling of R.roxburghii fruit during ripening
During ripening ofR.roxburghii,the growth rate of the fruit was slower in the early stage,and the fruit expanded rapidly and turned yellow in color when it entered the fruit expansion period (Fig.1-A). In addition,previous studies have shown that the total flavonoid content ofR.roxburghiifruit significantly accumulates during fruit development (Fanet al.2021). To investigate the dynamics of metabolites duringR.roxburghiifruit ripening,we performed broadly targeted LC-tandem MS (LC-MS/MS)-based metabolomic and transcriptomic sequencing using samples from the five stages ofR.roxburghiidevelopment (Fig.1-A). A total of 1,138 compounds were identified in the five fruit stages,with flavonoids being the most prevalent (287) and lignans and coumarins being the least prevalent (43). Principal component analysis (PCA)was carried out to cluster all the compounds detected by LC-MS/MS. This analysis revealed that all biological replicates were clustered together,indicating a high degree of confidence in the metabolomic data (Fig.1-B).We then clustered the samples based on transcriptomic data,which also resulted in the biological replicates clustering together,further demonstrating the reliability of this data (Fig.1-C). In addition,in a PCA analysis of the metabolome and transcriptome,PC1 values were 41.75 and 41.68%,and PC2 values were 29.57 and 14.32%,respectively. These results suggest that there are differences in the metabolites and genes during the ripening ofR.roxburghiifruit.
3.2.ldentification of differentially accumulated metabolites and differentially expressed genes during R.roxburghii fruit development

Fig. 1 Rosa roxburghii fruit at different sampling times,and principal component analysis of the metabolome and transcriptome. A,phenotype of R.roxburghii at different developmental periods. B and C,principal component analysis of the five samples based on metabolite contents and gene expression. S1,S2,S3,S4,and S5 represent 10,40,70,100 and 130 days after anthesis,respectively.
Differential accumulation of metabolites between samples(S1vs.S2,S2vs.S3,S3vs.S4,and S4vs.S5) was determined based on two criteria: VIP≥1 and FC≥1. Many metabolites were found to differentially accumulate duringR.roxburghiifruit development,including 537,192,141,and 571 different compounds in the S1vs.S2,S2vs.S3,S3vs.S4,and S4vs.S5 comparisons,respectively(Appendix B). We then analyzed the transcriptome data of the five fruit samples. The total filtered reads from each sample ranged from 5.69 to 8.22 Gb,the Q30 ranged from 91.00 to 94.31%,and the alignment rates to the genome ranged from 95.55 to 97.27%. There were totals of 3,819,7,892,1,047,and 733 differentially expressed genes in the S1vs.S2,S2vs.S3,S3vs.S4,and S4vs.S5 comparisons,respectively (Appendices C–E).
To identify the KEGG pathways that were enriched in both the transcriptome and metabolome data,KEGG enrichment analyses were performed for the differentially accumulated metabolites and the differentially expressed genes,with the top 25 pathways retained based on theP-values from the transcriptome enrichment analysis (Fig.2-A–D). The KEGG pathways commonly enriched in all samples were flavonoid biosynthesis,phenylpropanoid biosynthesis,ABC transporters,anthocyanin biosynthesis,and cyanoamino acid metabolism. We further analyzed the differentially accumulated metabolites in the four groups of samples(Fig.2-E),and found a total of 21 core components that were differentially accumulated in all stages. These components included two amino acids and derivatives,seven flavonoids,one lignan,one coumarin,two lipids,three organic acids,one phenolic acid,three terpenoids,and two other components (Fig.2-F).
3.3.Co-expression network analysis and candidate gene identification
We divided the flavonoids into five clusters. Among them,the compounds of cluster 5 were positively correlated withR.roxburghiifruit development and peaked at fruit ripening (Fig.3-A;Appendix F). We then performed an iterative screening of all genes using WGCNA with the flavonoid content in cluster 5 serving as the phenotypic data. The results of this analysis indicated that numerous flavonoid substances were positively correlated with MEblue,MEgreen,and MEpink,and negatively correlated with MEbrown and MEturquoise (Fig.3-B,Appendix G).
In addition,13 out of 39 components identified were quercetin derivatives (Appendix F). Quercetin and glycoside compounds are typical flavonoids,with anti-tumor,anti-viral,anti-diabetic,and cardiovascular protective effects (Wanget al.2022). We selected four of the quercetin derivatives,quercetin-3-O-arabinoside(guaijaverin),quercetin-3-O-galactoside (hyperin),quercetin-3-O-glucoside (isoquercitrin),and quercetin-7-O-glucoside,for further analysis. The correlations between modules and quercetin derivatives were visualized using the ggcor R package (Fig.3-C). Among the 10 modules,guaijaverin showed significant positive correlations with the magenta,green,and blue modules,withr2values of 0.73,0.61,and 0.90,respectively.Hyperin,isoquercitrin,and quercetin-7-O-glucoside showed positive correlations with the pink,green,and blue modules,withr2values ranging from 0.69 to 0.95. Furthermore,these four compounds showed negative correlations with MEturquoise (Fig.3-C). We also identified the genes in the biosynthetic pathway of quercetin and its derivatives,and found thatRrCHI,RrFLS,RrF3H,andRrF3´Hwere negatively correlated with the turquoise and brown modules,whereas three UDP-glycosyltransferases were positively correlated with the blue module.
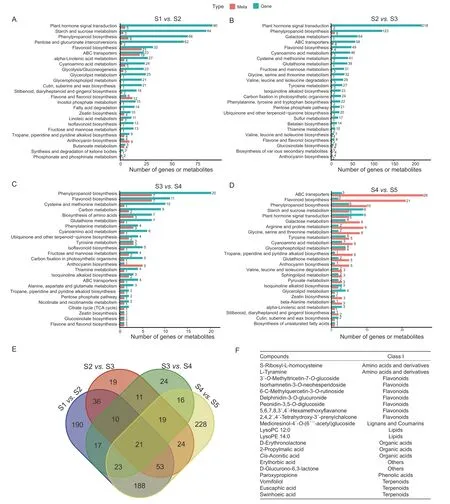
Fig. 2 Identification and functional characterization of differentially accumulated metabolites (DAMs) and differentially expressed genes (DEGs) in Rosa roxburghii fruit at different stages. A–D,KEGG enrichment analysis of the DAMs and DEGs between S1 vs.S2 (A),S2 vs.S3 (B),S3 vs.S4 (C),and S4 vs.S5 (D). E,Venn diagram depicting metabolites in the comparison of the four fruit samples. F,list of 21 core differentially accumulated metabolites among the five stages of fruit samples. S1,S2,S3,S4,and S5 represent 10,40,70,100 and 130 days after anthesis,respectively.

Fig. 3 Dynamics of flavonoid content and weighted gene co-expression network analysis (WGCNA). A,dynamics of the flavonoid content during R.roxburghii fruit ripening. B,module–flavonoid relationships. Each row represents a module,and each column represents a metabolite. C,correlation of modules with quercetin derivatives. S1,S2,S3,S4,and S5 represent 10,40,70,100 and 130 days after anthesis,respectively.
To investigate the regulatory network of quercetin and its derivatives inR.roxburghii,we reconstructed the biosynthetic pathways of quercetin and its derivatives,and analyzed the expression profiles of related gene family members. We found that the precursor compounds and genes involved in the biosynthesis step from naringenin chalcone to quercetin and its derivatives were divided into two accumulation patterns (Fig.4). One group of precursor compounds and related genes involved in the synthesis of quercetin decreased during fruit development and reached their lowest values at fruit ripening. The other group was composed of quercetin derivatives and related biosynthetic genes that were upregulated during fruit development and peaked at fruit ripening (Fig.4).These results suggest thatRrUGT73BandRrUGT78Dare key genes involved in the synthesis of quercetin derivatives.
3.4.ldentification of transcription factors regulating the synthesis of quercetin derivatives in R.roxburghii
To validate the role of transcription factors in regulating the synthesis of quercetin derivatives inR.roxburghii,we selected three UDP-glycosyltransferases in the blue module (evm.TU.chr2.83,evm.TU.chr4.5103,andevm.TU.chr4.5104) for further analysis. The expression profiles of all genes in the blue module were analyzed(Appendix H),and all the upregulated genes from this module were annotated using blast and hmmscan. A total of 178 transcription factors were found to be upregulated over the course of fruit development (Appendix I). These genes encode various types of transcription factors,including bHLH,bZIP,NAC,WRKY,and others,which may have important roles in regulating the synthesis of quercetin derivatives inR.roxburghii. According to the FPKM>10 and >two-fold criteria,we screened 16 highconfidence transcription factors from the 178 transcription factors,includingRrNAC042,RrbZIP6,RrWRKY4,RrGBF3,RrANL2,RrPIF8,RrHY5-homolog,RrBZIP9,RrARF17,RrWOX13,RrMYBS1,RrLZF1,RrBEH4,RrLBD1,RrGRAS2,andRrWRKY44(Appendix J).

Fig. 4 Metabolic pathways of quercetin and its derivatives in Rosa roxburghii. Histograms represent metabolite contents,and heat maps represent gene expression levels. CHI,chalcone isomerase;F3H,flavanone 3-hydroxylase;F3´H,flavonoid 3´-hydroxylase;FLS,flavonol synthase;UGT73B,flavonol-7-O-glucosyltransferase;UGT78D,flavonol-3-O-rhamnosyltransferase. S1,S2,S3,S4,and S5 represent 10,40,70,100 and 130 days after anthesis,respectively.
To confirm the relationships between these 16 transcription factors and UDP-glycosyltransferase,the interactions between the transcription factors and the promoters were verified by yeast one-hybrid assays. The results revealed that RrPIF8 could bind to the promoter of UDP-glycosyltransferaseevm.TU.chr2.83;RrBEH4 and RrWOX13 could bind to the promoter ofevm.TU.chr4.5013;and RrLBD1,RrPIF8,and RrMYBS1 could bind to the promoter ofevm.TU.chr4.5014(Fig.5-A). We used dual luciferase assays to further confirm the binding of transcription factors to these promoters. The results indicated that RrPIF8 had no effect on the fluorescence signal of luciferase driven by the promoter ofevm.TU.chr2.83. RrBEH4 promoted the fluorescence signal of pro-evm.TU.chr4.5013::LUC,whereas RrWOX13 inhibited the fluorescence signal of this construct. RrLBD1 and RrPIF8 could bind to pro-evm.TU.chr4.5014 and increase the LUC signal intensity,whereas RrMYBS1 co-expressed with pro-evm.TU.chr4.5014::LUC had no significant effect on the LUC signal intensity (Fig.5-B). Except for RrWOX13,we further confirmed these findings in tobacco leaves,and similar results were obtained (Fig.5-C). Finally,we further verified the expression patterns of three UDPglycosyltransferases,RrBEH4,RrLBD1andRrPIF8,duringR.roxburghiifruit development. The results showed that all the genes were significantly expressed at the S5 stage(Fig.6). These results suggest that these genes have important roles in the synthesis of quercetin derivatives inR.roxburghiifruit.
4.Discussion
The fruit ofR.roxburghiiis rich in ascorbic acid,flavonoids,vitamins,and other compounds that are beneficial to human health (Wanget al.2021;Liet al.2022). Consumption ofR.roxburghiihas been shown to improve immunity,slow aging,lower blood pressure,and lower blood lipid levels (Aburtoet al.2013). It has also been found to have anti-cancer properties(Heet al.2016),liver-protective qualities (Huanget al.2014),and atherosclerosis-preventative effects (Zhanget al.2001). Flavonoids are one of the most important groups of bioactive compounds inR.roxburghiifruit,and our metabolomic analysis revealed dynamic changes in 287 flavonoids over the course ofR.roxburghiifruit development,with flavonoid levels peaking at the ripening stage. The levels of quercetin derivatives were found to increase significantly during ripening,and these compounds have been found to promote human health without any known side effects. For instance,quercetin and its derivatives positively impact human health in a variety of ways through their anti-tumor,antiaging,hypotensive,hypoglycemic (Jeonget al.2012),and hypolipidemic (Lee and Mitchell 2011) effects. High quantities of enriched quercetin derivatives may be responsible for the medicinal benefits ofR.roxburghii.Therefore,understanding the molecular mechanisms underlying the accumulation of quercetin and its derivatives could lead to the creation of newR.roxburghiicultivars with improved health benefits.
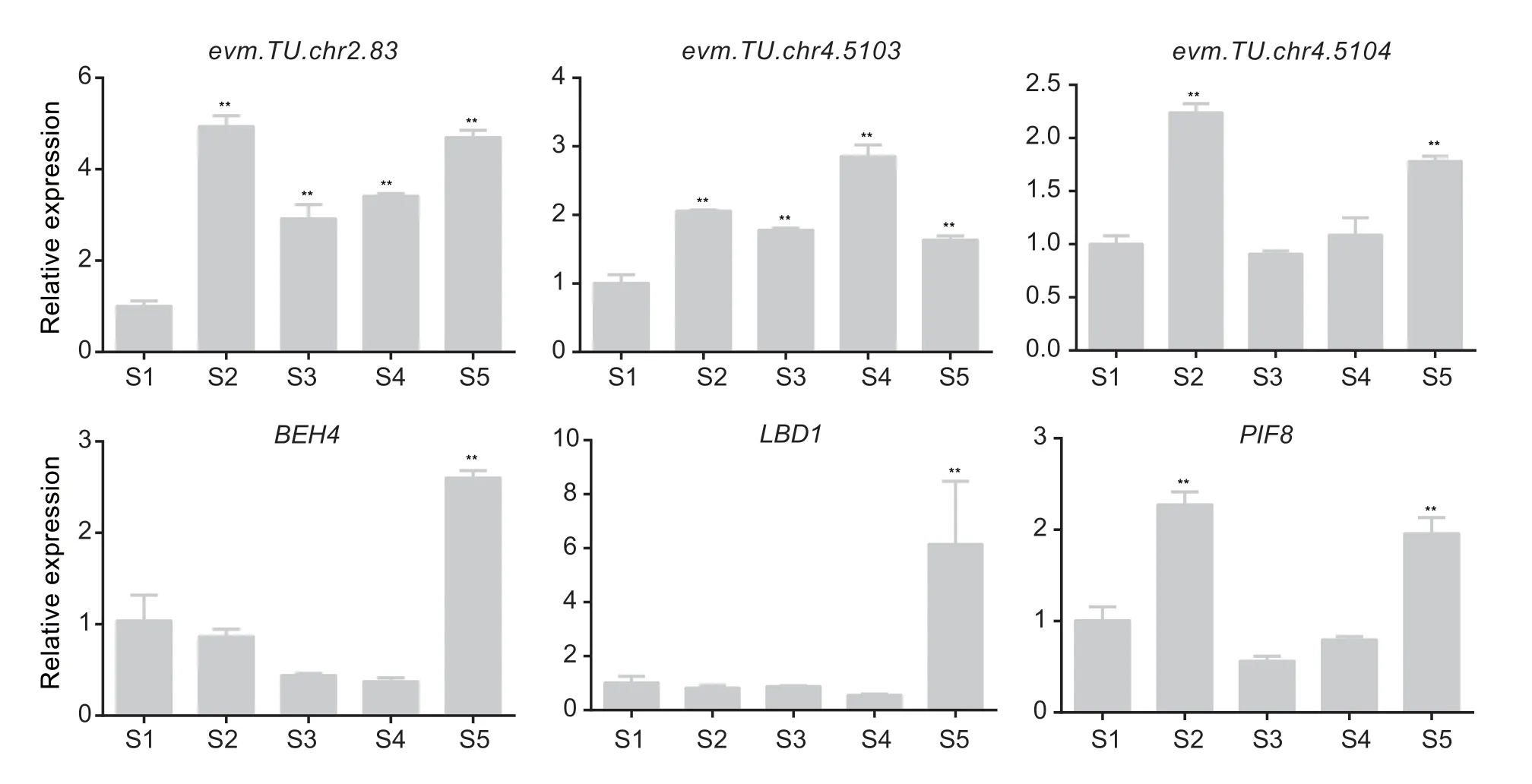
Fig. 6 Relative expression of six genes involved in quercetin derivative metabolism during Rosa roxburghii fruit development. S1,S2,S3,S4,and S5 represent 10,40,70,100 and 130 days after anthesis,respectively. Asterisks indicate significant differences(LSD,P<0.01).
The anabolism of flavonoids is a complex biological process that is regulated by multiple factors. We constructed correlation-based models to identify the relevant gene regulatory networks. This approach has previously been applied to horticultural crops such as watermelon (Umeret al.2020),mango (Wuet al.2022),kiwifruit (Wanget al.2022),and pepper (Liuet al.2020).In this study,we used the WGCNA approach to integrate the transcriptomic and metabolomic data inR.roxburghii,for the first time,and identified important genes involved in the biosynthesis of quercetin derivatives.Furthermore,our data showed that quercetin and its precursors accumulated in the early and middle stages ofR.roxburghiifruit development,but the levels of these compounds decreased significantly during fruit ripening.As quercetin levels declined,its derivatives significantly increased over the course ofR.roxburghiifruit ripening.Additionally,the genes related to the synthesis of quercetin and its precursors showed similar trends.Quercetin is inherently unstable in plants,and is typically found in the form of quercetin glycosides. We identified two genes related to the synthesis of quercetin derivatives inArabidopsisthaliana,UDP-GLUCOSYLTRANSFERASE 73B2 (Joneset al.2003) and UDP-glucosyl transferase 78D2 (Limet al.2002),which were found in the gene expression modules that were significantly correlated with quercetin levels. These genes served as hub genes for the modules,and their expression profiles were consistent with changes in the levels of the quercetin derivatives.
In addition to the structural genes directly involved in flavonoid biosynthesis,transcription factors also play a key role in regulating the synthesis of flavonoids,but their roles in controlling the levels quercetin derivatives have not been sufficiently investigated (Mehrtenset al.2005;Strackeet al.2007;Xuet al.2014;Liuet al.2015;Maet al.2018;Zhouet al.2019). Therefore,we comprehensively analyzed the transcription factors found in the gene expression modules that were significantly associated with quercetin derivatives,and found that many transcription factors were positively associated with the accumulation of these derivatives. We selected 16 of these transcription factors for yeast one-hybrid and dual luciferase assays. The results indicated that RrBEH4,RrLBD1,and RrPIF8 could significantly increase the expression ofRrUGT73B2andRrUGT78D2,which regulate the synthesis of quercetin derivatives inR.roxburghii. Additionally,BEHis a key transcription factor in brassinolide signaling (Wanget al.2002;Yinet al.2005),indicating that brassinolides may be involved in the biosynthesis of quercetin derivatives inR.roxburghii.LDBis a plant-specific gene family,whose members are differentially expressed in roots,leaves,flowers,and other organs (Borghiet al.2007;Soyanoet al.2008;Yordanovet al.2010). An earlier study found that the LBD family is involved in flavonoid synthesis (Songet al.2020),and we found thatLBD1regulates the synthesis of quercetin derivatives inR.roxburghii. PIFs are a class of lightresponsive bHLHs,andPIF8has been shown to play a role in the light response (Ohet al.2020). These findings imply that quercetin derivative synthesis may be influenced by light exposure inR.roxburghii. Taken together,our results indicate that quercetin derivative accumulation is affected by hormones,light,and other factors,although additional work is needed to understand the various factors that influence their metabolism inR.roxburghii.
5.Conclusion
In this work,we investigated the dynamic changes of flavonoids and the regulation of the synthesis of quercetin derivatives duringR.roxburghiifruit development using transcriptomic and metabolomic approaches. Quercetin derivatives were found to accumulate to high levels duringR.roxburghiifruit ripening,which likely impacts its medicinal properties. In addition,we identified several structural genes in the biosynthetic pathways of quercetin derivatives as well as key enzymes and regulatory factors that affect the synthesis of quercetin derivatives,laying a foundation for the further improvement ofR.roxburghiifruit.
Acknowledgements
This research was supported in part by the Priority Academic Program Development of Jiangsu Higher Education Institutions and the State Key Laboratory of Crop Genetics and Germplasm Enhancement,China(ZW201813). This study was also supported by the highperformance computing platform of the Bioinformatics Center,Nanjing Agricultural University,China.
Declaration of competing interest
The authors declare that they have no conflict of interest.
Appendicesassociated with this paper are available on https://doi.org/10.1016/j.jia.2023.11.022
 Journal of Integrative Agriculture2024年3期
Journal of Integrative Agriculture2024年3期
- Journal of Integrative Agriculture的其它文章
- Molecular mechanisms of stress resistance in sorghum: lmplications for crop improvement strategies
- Artificial selection of the Green Revolution gene Semidwarf 1 is implicated in upland rice breeding
- Dynamics and genetic regulation of macronutrient concentrations during grain development in maize
- The NAC transcription factor LuNAC61 negatively regulates fiber development in flax (Linum usitatissimum L.)
- The underlying mechanism of variety–water–nitrogen–stubble damage interactions on yield formation in ratoon rice with low stubble height under mechanized harvesting
- Rice canopy temperature is affected by nitrogen fertilizer