
Genome-wide association study of seedling nitrogen-use efficiency-associated traits in common wheat (Triticum aestivum L.)
2024-03-07 01:50HuweiShiWeichongWngLifengGoJirongWuChengmeiHuHuishuYnYugngShiNingLiYouzhiYonginZhouZhoshiXuJunChenWensiTngKiChenDizhenSunYuxingWuMingChen
The Crop Journal 2024年1期
Huwei Shi, Weichong Wng, Lifeng Go, Jirong Wu, Chengmei Hu, Huishu Yn, Yugng Shi,Ning Li, Youzhi M, Yongin Zhou, Zhoshi Xu, Jun Chen, Wensi Tng, Ki Chen, Dizhen Sun,*,Yuxing Wu,*, Ming Chen,*
aCollege of Agriculture, Shanxi Agricultural University, Jinzhong 030801, Shanxi, China
b National Key Facility for Crop Gene Resources and Genetic Improvement,Key Laboratory of Biology and Genetic Improvement of Triticeae Crops,Ministry of Agriculture,Institute of Crop Sciences, Chinese Academy of Agricultural Sciences, Beijing 100081, China
Keywords:Wheat NUE Genome-wide association study Nitrogen sensitive index Chlorate inhibition rate
ABSTRACT Nitrogen(N)fertilizer application is essential for crop-plant growth and development.Identifying genetic loci associated with N-use efficiency(NUE)could increase wheat yields and reduce environmental pollution caused by overfertilization.We subjected a panel of 389 wheat accessions to N and chlorate(a nitrate analog)treatments to identify quantitative trait loci(QTL)controlling NUE-associated traits at the wheat seedling stage.Genotyping the panel with a 660K single-nucleotide polymorphism(SNP)array,we identified 397 SNPs associated with N-sensitivity index and chlorate inhibition rate.These SNPs were merged into 49 QTL, of which eight were multi-environment stable QTL and 27 were located near previously reported QTL.A set of 135 candidate genes near the 49 QTL included TaBOX (F-box family protein) and TaERF(ethylene-responsive transcription factor).A Tabox mutant was more sensitive to low-N stress than the wild-type plant.We developed two functional markers for Hap 1, the favorable allele of TaBOX.
1.Introduction
Nitrogen(N)is an essential nutrient for crop growth and development.As the natural supply of N in the soil is often insufficient to meet crop demand,high crop yields depend heavily on N fertilizer application [1].They also depend on crop N-use efficiency(NUE), with only one third of applied N used by wheat plants [2].A better understanding of plant N uptake and transportation is desirable for breeding crops with high NUE [3].Knowledge of the biological mechanisms underlying plant NUE is obtained mostly from model plant species such as Arabidopsis and rice.Owing to the complexity of its genome, limited information is available about NUE genetics in common wheat.
Improving NUE is challenging because of the complexity of plant mechanisms for responding to N deficiency.N-deficiency responses encompass N uptake, transport, reduction, assimilation,translocation,and remobilization[2].A study of nitrate signaling is one route to NUE improvement.Nitrate absorption and transport by higher plants is performed mainly by members of the nitrate transporter 1/peptide transporter family (NPF).Of at least 53 NPF genes identified in Arabidopsis, 19 are involved in nitrate uptake and transport [4].In 2022, respectively 92 and 88 NPF genes were identified in foxtail millet (Setaria italica L.) and its wild ancestor green foxtail (S.viridis L.) [5].Transcription factors (TFs) function in the regulatory network underlying NUE.The NAM, ATAF, and CUC (NAC) families, which are the main TF families for NUE [6].ANR1 was the first reported MADS-box TF with a role in the nitrate-signaling pathway and has been shown [7] to regulate lateral root production,by regulating nitrate transporter 1.1(NRT1.1)in Arabidopsis.Various TFs of the NIN-like protein(NLP)family are also involved in N uptake and associated signaling pathways [8],such as AtNLP7, a major regulator involved in nitrate-responsive gene activation in Arabidopsis [9].Recent study [10] have shown that the NLP7 protein is not only a TF of the nitrate signaling pathway but also an intracellular nitrate sensor and receptor.Further characterization of TFs in wheat might provide clues to the mechanisms regulating NUE.
Genome-wide association study(GWAS)is a promising method for characterizing the regulatory networks of NUE in crops.In a recent study [11], 3,356,591 single-nucleotide polymorphism(SNP) sites were tested for association with flag leaf width (LW)in 134 rice varieties, with LW positively correlated with N stress.Forty-two quantitative trait loci (QTL) were associated with flag LW under various N treatment conditions.In a GWAS of 373 soybean varieties genotyped for 31,145 SNPs, respectively 17, 19,and 24 SNPs were associated with N, N concentration, and the Cto-N ratio[12].In another GWAS using a diverse panel of 230 rice accessions, 411 genes located in five QTL regions and 2722 differentially expressed genes were identified in response to low N(LN)supply [13].
Because plants have the same mechanisms of absorption of chlorate and nitrate, N absorption efficiency can be measured as chlorate tolerance.The AtNRT1.1B[14]and OsNRT1.1B[15]nitrate transporters have been cloned from Arabidopsis and rice and can be used for simulating plant nitrate absorption and phenotype identification [16,17].
We have conducted a GWAS for eight NUE-associated agronomic traits of wheat plants at the maturity stage[18].Identifying QTL associated with NUE-associated traits under field conditions is subject to long experimental cycles and environmental effects.Although it may be easier to phenotype seedlings than adult plants[19],the pathways controlling NUE at the two stages may differ.In the present study, we aimed to identify QTL controlling NUErelated traits in the wheat seedling stage using the same GWAS panel as that used in our previous study.
2.Materials and methods
2.1.Populations and genotypic datasets
A panel of 389 winter wheat cultivars and elite breeding accessions were collected from the major wheat-producing regions of China.Based on the population structure, all accessions were classified into two subgroups as previously described [18].
2.2.Phenotyping of common wheat NUE at the seedling stage
Two independent hydroponic culture trials were conducted at Shanxi Agricultural University: E1 from April 26 to September 30,2021 and E2 from June 20 to December 11,2022.Two N treatments—LN and high N (HN)— were applied, each repeated three times.Wheat seeds were surface-sterilized with 2% H2O2for 30 min and germinated in distilled water.After 3 d,the seedlings were transferred to a 96-well black plastic box and cultured in a light incubator under 14 h light:10 h darkness.The temperature was maintained at 22 °C during the light period and at 18 °C during the dark period.N concentrations in the Hoagland nutrient solutions were 0.2 (LN) and 2 mmol (HN) L-1and concentrations corresponded to those of the N source Ca(NO3)2∙4H2O, which was supplied at 0.1 and 1 mmol L-1.The solution was replaced every 3 d.
After 3 weeks of growth, for each treatment of each variety,seven plants were selected at random and their plant height(PH),root length(RL),leaf length(LL),leaf width(LW),and number of roots (RN) were recorded.Each plant was separated into stem and roots and root fresh weight(RFW)and seedling(stem and leaf)fresh weight(SFW)were measured.Plants were then killed at 105°C for 30 min and dried at 70 °C to constant weight.Root dry weight (RDW) and seedling dry weight (SDW) were recorded.
Plant fresh weight (PFW) = RFW + SFW
Plant dry weight(PDW)=RDW+SDW Root/shoot ratio(RSR)=RDW/SDW
The chlorate trial lasted from January 12 to March 5, 2022.Wheat seedlings were grown as described above.After 7 d of growth (at the two-leaf stage), seedlings in a control group were cultured in modified Hoagland nutrient solution and those in a chlorate-treated group in a nutrient solution containing 4 mmol L-1KClO3instead of 1 mmol L-1Ca(NO3)2.After 4 d of further growth, the heights of seedlings were recorded, and the chlorate inhibition rate (CIR) of plant growth was calculated using the following formula: CIR = (control PH - chlorate PH)/control PH × 100%.Each treatment was repeated three times, using four seedlings each time.SPSS 21.0 (IBM, Armonk, NY, USA) was used to calculate correlations among traits and compare treatment mean phenotypes by t-test.Broad-sense heritabilities(H2)of traits were estimated as previously described [20].
2.3.GWAS
The 389-accession panel was genotyped using the high-density wheat 660K SNP array.A total of 397,384 high-quality SNPs with minor-allele frequency > 0.05 and missing data rate < 20% were used for GWAS [18].
Best linear unbiased prediction(BLUP)values for each accession across the two environments (three replicates per environment)were calculated using a linear model with random effects for variance components,using the LME4 package in R[21].A N sensitivity index (NSI) was calculated following Fischer and Maurer [22] for PH (PHR), RL (RLR), RN (RNR), LL (LLR), FSN (LWR), RFW (RFWR),SFW (SFWR), RDW (RDWR), SDW (SDWR), RSR (RSRR), PFW(PFWR), and PDW (PDWR).The GWAS was performed using a mixed linear model (MLM) with GAPIT 3.0 [23].The NSI for each trait in both environments studied, BLUP values across both environments, and CIR values were used in GWAS.
The suggested genome-wide P-value threshold of P=1/Ne,where Ne represents the effective number of SNPs calculated using the Genetic Type 1 Error Calculator[24](Table S1)was 1.49×10-5.To eliminate false negatives caused by an excessively strict Bonferroni correction,an unadjusted P-value<1×10-4was used for the GWAS of CIR.Associated markers and P-value distributions were visualized with Manhattan and quantile–quantile (Q–Q) plots drawn with the qqman package in R 3.6.1 (http://www.r-project.org/).The phenotypic variance (R2) explained by SNPs was calculated with GCTA [25].The GOseq R package [26] was used to perform Gene Ontology (GO) analysis of candidate genes.KOBAS 2.0[27] was used to perform Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis.
2.4.Linkage disequilibrium (LD) analysis, QTL identification, and candidate gene annotation
SNPs in the 2.0-kb promoters region of candidate genes and nonsynonymous SNPs in the coding sequences were used for haplotype analysis.The SNPs in LD blocks with r2≥0.5 were merged as a single QTL.The LD between SNPs was calculated with TASSEL 5.0[28,29].Pairwise LDs of SNPs within each QTL were visualized using the R package LDheatmap [29].Candidate genes were screened from all annotated genes according to the following criteria:(1)homologous genes in Arabidopsis or rice were identified in a previous study as being involved in N and senescence metabolism,and (2) genes were upregulated or downregulated after LN treatment.The results from our previous study[30]of the LN transcriptome were used to screen for candidate genes.
2.5.RNA extraction and gene expression quantification
To investigate the expression patterns of candidate genes following short-term nitrate treatment, we grew 7-d-old seedlings of the Chinese variety Shimai 15 in a nutrient solution containing 1 mmol L-1Ca(NO3)2.N was withheld for 2 d before the plants were placed in fresh solution for 48 h [31].
Seven-day-old seedlings of five High-PDWR, five Low-PDWR,five High-CIR, and five Low-CIR accessions were grown for 14 d under long-term treatment conditions.The seedlings were placed in nutrient solution containing either 1 mmol L-1Ca(NO3)2(HN)or 0.1 mmol L-1Ca(NO3)2(LN)[32].Each treatment was replicated three times, and samples of both shoots and roots were collected.Total RNA was extracted with a Total RNA Extraction Kit(TIANGEN,Beijing, China).cDNA was synthesised from total RNA using the ReverTra Ace qPCR RT Master Mix (Toyobo, Kyoto, Japan).Quantitative reverse transcription PCR was performed using the SYBR Green Real-time RT-PCR kit (Takara, Kyoto, Japan).Relative gene expression was estimated by the 2-ΔΔCTmethod [33].The experiment was conducted in triplicate.
2.6.Functional marker development and haplotype network analysis
Kompetitive Allele-Specific PCR (KASP) primers for KASP-SNP2 and KASP-SNP7 (Table S7) were designed using the WheatOmics v1.0 website (http://wheatomics.sdau.edu.cn) [34].The reaction volume of each assay was 5 μL, consisting of 100 ng μL–1DNA,2.5 μL of 1×KASP master mixture,0.04 μL MgCl2,0.056 μL primer mix (0.012 μL of each forward A and B primers and 0.031 μL of reverse primer),and 0.205 μL ddH2O.Amplification was performed with an ABI Veriti 384 PCR instrument (Thermo Fisher Scientific,Waltham, MA, USA) under the following conditions: 94 °C for 15 min, followed by 10 touchdown cycles (94 °C for 15 s; 61 °C initially and decreasing by 0.6 °C per cycle for 20 s), and 29 cycles of 94 °C for 20 s and 57 °C for 60 s.
The haplotypes of candidate genes were constructed from highquality whole-genome and SNP data of wheat(Wheat SnpHub Portal: http://wheat.cau.edu.cn/Wheat_SnpHub_Portal/).The dataset contained resequencing data of 681 hexaploid wheat varieties published since 2019 [35].
2.7.Phenotypic identification of Tabox mutants generated in response to N stress
A search of the Ethyl Methane Sulfonate (EMS)-induced Wheat Jing 411 TILLING Database (http://jing411.molbreeding.com/)identified Tabox(4B_667 814 715_E)as a homozygous EMS mutant with STOP_GAINED mutation sites for Tabox.The mutation sites were validated by sequencing the PCR products amplified by specific primers (Table S7).Two N treatments (LN and HN) were used for identifying Tabox mutants.The experimental conditions were similar to those described in section 2.2, imposing N treatments after 3 weeks of growth.
3.Results
3.1.Correlations among NUE-associated agronomic traits at seedling stage
Traits recorded for treated plants(Figs.S1–S3)showed continuous variation under both N conditions and control and chlorate treatments.All traits but RL and RSR were increased under HN.Genotype, treatment, and their interaction G × T effects were significant for all traits.Coefficients of variation (CV) ranged from 15.36% for RN to 78.25% for RFW under HN conditions and from 13.71% for RN to 88.60% for RFW under LN conditions.The CV of both the RFW and RDW were relatively high under both N treatments.The H2of agronomic traits was moderate to high, varying from 0.46 for RSR to 0.89 for RFW under HN conditions and from 0.68 for PFW to 0.91 for RDW under LN conditions.However,RSR and PH had lower H2than the other agronomic traits under HN and LN conditions.Descriptive statistics for these traits are presented in Table S2.
PH was negatively correlated with all traits but RL and LL,under HN conditions (Fig.S4A).Likewise, LL was negatively correlated with all traits except RL and PH.The strongest correlations were observed among RFW,SFW,RDW,SDW,PFW,and PDW.The correlations among agronomic traits under LN conditions were similar to those under HN conditions (Fig.S4B).RSRR was negatively correlated with all traits but RDWR,RFWR,and RLR.Other traits were significantly positively correlated with one another (Fig.S4C).
In the chlorate inhibition experiment, the maximum seedling height was 26.66 cm, the minimum was 9.94 cm, the mean was 17.64 cm, and the CV was 16.59% under control conditions(Table S3).After treatment with 4 mmol L-1potassium chlorate,the seedling heights of wheat varieties in the natural population were lower than those observed under control conditions.After 4 d of potassium chlorate treatment, the maximum seedling height was 20.54 cm, the minimum was 7.67 cm, the mean was 12.48 cm, and the CV was 17.24%.The mean seedling height differed between the control and chlorate treatments.The highest CIR for seedlings across these varieties was 53.93%, the lowest was 2.15%, the mean was 28.77%, and the CV was 29.24% (Table S3).
3.2.Marker–trait associations and QTL analysis revealed candidate genes and pathways related to NUE in wheat
Marker-trait associations (MTAs) identified using the MLM model are shown in Table S4 and Figs.1,S5,S6.The Q–Q plots indicated proper control of false positives in this GWAS (Figs.S7–S9).GWAS results for the studied traits are described in Table S4.A total of 397 MTAs were identified for the 13 studied traits from the E1 and E2 environments and BLUP estimates(Table S3).Based on the LD between significant SNPs,these MTAs were combined to form 49 QTL:3 for RLR,5 for SDWR,2 for RDWR,3 for SFWR,4 for RFWR,1 for PHR,6 for RSRR,5 for LLR,3 for LWR,4 for PFWR,7 for PDWR, and 6 for CIR.The QTL intervals ranged from 0.008 to 7.2 Mb.Among them, 12 QTL, including qLLR3A.1, qLLR3A.2,qPFWR5B.1, qRDWR2A.1, qRFWR2B.1, qRFWR5A.1, qRLR2B.1,qRSRR3B.1,qSDWR3B.1,qSFWR5B.1,qCIR1A.2,and qCIR1A.5,showed≤ 1.0-Mb intervals.Individual SNPs explained 4.7% of AX-109319594 for qLWR1A.1 and 7.2%of AX-108846679 for qPFWR5B.1 in the phenotypic variation.qSFWR5B.1 overlapped with qPFWR5B.1, qRDWR2A.1 overlapped with qSDWR2A.1 and qRFWR2A.1, and qPDWR4B.1.overlapped with qPFWR4B.1 and qSFWR4B.1.Most QTL were detected in only a single environment,whereas eight QTL were detected in at least two environments:qLLR3A.1, qLLR3A.2, qLWR3A.1, qPDWR4B.1, qPHR4A.1, qRDWR2A.1,qRFWR2B.1, and qRLR2B.2.
Of the 49 QTL, 27 were located near previously mapped NUE QTL (Table S4).A total of 1351 genes in the 49 QTL regions were annotated, with 0–99 genes per QTL (Table S5).The GO analysis of candidate genes revealed enrichment in multiple biological processes, including ‘‘reactive nitrogen species metabolic process”,‘‘regulation of nitrogen compound metabolic process”, ‘‘cellular nitrogen compound biosynthetic process”, and ‘‘nitrogen compound metabolic process” (Fig.S10).Candidate genes involved in the N metabolism pathway were identified via KEGG pathway analysis.TraesCS6A02G017500 in qLLR6A.1 was involved in N metabolism; it was annotated as nitrate reductase and functions in nitrate assimilation.TraesCS2B02G368400, TraesCS2B02G368500,TraesCS2B02G368600, and TraesCS2B02G368700 in qRSRR2B.1 were annotated as nitrate transporter 1.2 genes, encoding inducible components of low-affinity nitrate uptake [36].
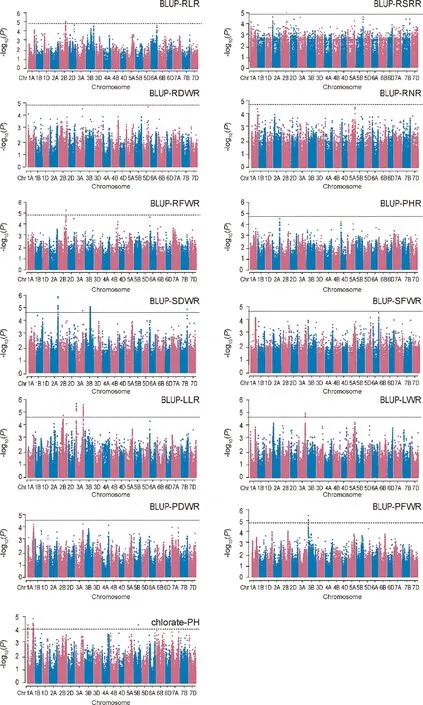
Fig.1.GWAS Manhattan plots for N sensitivity index (NSI) from BLUP estimate and chlorate inhibition rate.Black solid horizontal lines in the Manhattan plots indicate thresholds.PHR refers to NSI of PH;RLR refers to NSI of RL;LLR refers to NSI of LL;RNR refers to NSI of RN;RFWR refers to NSI of RFW;SFWR refers to NSI of SFW;RDWR refers to NSI of RDW;SDWR refers to NSI of SDW;PFWR refers to NSI of PFW;PDWR refers to NSI of PDW;RSRR refers to NSI of RSR;BLUP,best linear unbiased prediction across the two environments; PH, plant height; RL, root length; LL, leaf length; LW, leaf width; RN, root number; RFW, root fresh weight; SFW, seedling fresh weight; RDW, root dry weight; SDW, stem dry weight; PFW, plant fresh weight; PDW, plant dry weight; RSR, root/shoot ratio; Chlorate-PH, PH under control and chlorate conditions.
3.3.Identification of potential N-responsive genes using homologybased screening and expression analysis
Based on the findings of our previous study on the LN transcriptome[30]and the functions of homologous genes,we selected 135 candidate genes from all annotated genes(Table S6).Among these,71 were either upregulated or downregulated in response to LN treatment.TraesCS6A02G018100, located at qLLR6A.1, encodes a mitochondrial transcription termination factor-like protein, with a log2(FC, fold change) of –8.79.TraesCS4B02G392900 (TaBOX),located at qPDWR4B.1 and encoding an F-box family protein box,showed the greatest upregulation after LN treatment, with a log2(FC) of 6.7.
Several candidate genes homologous to those in Arabidopsis have functions associated with N mobilization, nitrate assimilation, N remobilization, and low nitrate-induced anthocyanin biosynthesis.Three genes encoding cysteine proteases(TraesCS4B02G389800, TraesCS4B02G389900, and TraesCS4B02G390100)are involved in N mobilisation during senescence and two genes (TraesCS4D02G359100 and TraesCS5A02G558200) encoding TFs are involved in nitrate assimilation.Four genes (TraesCS1A02G331100, TraesCS1A02G292900, TraesCS4A02G439000, and TraesCS4A02G439400) N remobilization during leaf senescence,13 are in low nitrate-induced anthocyanin biosynthesis,and 10 in N assimilation.The presence of genes involved in N response in the regions of SNP–trait association confirmed the potential effects of NUE in wheat.
3.4.Identification of stable QTL and candidate genes for improving NUE in wheat
One of the eight multi-environment stable QTL detected was qPDWR4B.1, which is a major and stable QTL for PDWR.An improvement-selective sweep region on chromosome 4B overlapped with qPDWR4B.1 (Fig.2A).We calculated FSTby comparing the results from cultivars released before and after 1970.The first green revolution in the 1960s–1970s resulted in the successful cultivation of semi-dwarf crops,but also caused a decline in PH and a reduction in plant NUE [18].The LD block for qPDWR4B.1 was approximately 3.62 Mb.The lead SNP AX-94557938 hereafter represents this significant QTL (P=2.58E-06).Eight SNPs were significantly associated with qPDWR4B.1, exhibiting strong linkage disequilibrium (Fig.2A).Of 77 genes located in the qPDWR4B.1 region;four showed increased and one descreased expression after LN treatment(Table S6).TaBOX was located 1.3 kb upstream of the peak SNP AX-94557938 and was expressed at high levels(log2(FC)=6.7), exhibiting the maximum upregulation fold after LN treatment.We assigned it as the most promising candidate gene among those identified in response to N stress.TaBOX encodes an F-box family proteinResequencing of TaBOX revealed four major haplotypes (Hap 1–Hap 4) in the 2.0 kb promoter region and coding sequence, with two (SNP-6 and SNP-7) located in the coding region.SNP-6 was a synonymous mutation, whereas SNP-7 was a nonsynonymous mutation resulting in the conversion of Pro to His (Fig.2B).Hap 1 exhibited the highest PDWR of the four three haplotypes, suggesting that Hap 1 is the favorable allele of TaBOX for stable PDWR in response to LN stress (Fig.2C).To further elucidate the functional mechanism of TaBOX,we conducted an extensive analysis of TaBOX polymorphisms using public data.The TaBOX sequence could be divided into 11 haplotypes, with amino acid differences present in the exon variant sequences.Three haplotypes showed relative frequency > 0.1.The favorable haplotype Hap 1 was more similar to Hap III in the public data, with Hap III having a haplotype relative frequency of 0.19 (Fig.S11).We created two KASP markers: KASP-SNP-2 was designed for the SNP at 667,677,779 bp and KASP-SNP-7 for the SNP at 667,678,732 bp.These markers distinguished Hap 1 and Hap 4 from Hap 2 and Hap 3 (Fig.2B, D).
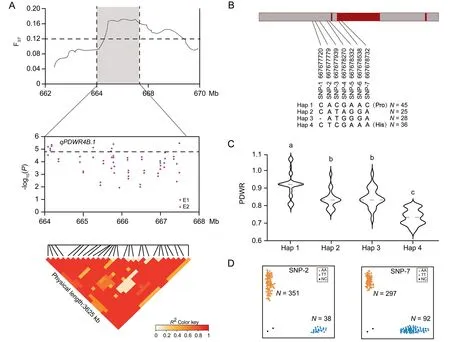
Fig.2.Variation in qPDWR4B.1 haplotypes controlling the N sensitivity index(NSI)of plant dry weight.(A)FST plot of cultivars released before and after 1970 across genomic regions surrounding the lead single-nucleotide polymorphism(SNP) AX-94557938 of qPDWR4B.1,PDWR:NSI of PDW.Local Manhattan plot and linkage disequilibrium (LD)heat map of SNPs within the LD block of qPDWR4B.1.E1,first trial;E2,second trial.(B)Four haplotypes(Hap 1,Hap 2,Hap 3,and Hap 4)were determined for TaBOX based on the variations of seven SNPs.Hap, haplotype.(C) Haplotype analysis of TaBOX.Violin plot showing the distribution of given traits for each haplotype.Different letters represent differences at P<0.05.(D)Kompetitive Allele-Specific PCR(KASP)assays for SNP-2 and SNP-7 of TaBOX in the 389 accessions.Scatterplots of KASP assays showing clustering of 389 accessions on the X-(FAM) and Y-(HEX) axes.
We identified 27 SNPs in the GWAS of CIR.The most significant SNP,AX-108739059(P=1.31E-05),was identified in QTL qCIR1A.3.The LD block of qCIR1A.3 was approximately 3.84 Mb(Fig.3A),and 15 significant SNPs were identified in this region.We also identified three haplotypes (Hap 1–Hap 3) based on 15 significant SNPs in the qCIR1A.3 region (Fig.3B), with Hap 3 contributing to a higher CIR than the other haplotypes (Fig.3C).The qCIR1A.3 sequence could be divided into seven haplotypes in the wheat panel.In our sequencing samples, the favorable haplotype Hap 3 was more similar to Hap IV in the public data, with Hap IV having a haplotype frequency of 0.49(Fig.S12).Of 47 genes located in the qCIR1A.3 region(Table S5),TraesCS1A02G328800(TaERF)was annotated as an ethylene-responsive TF, while its homolog AT3G23240 is reportedly [37] involved in the ERFs-NRT1.8 signaling module,which promotes NRT1.8-mediated nitrate unloading from the hypocotyl to roots and leaves.Speculating that TaERF functions in N assimilation and remobilization in wheat, we assigned TaERF as the most promising candidate gene for qCIR1A.3.
3.5.Expression of candidate genes
Nitrate treatment rapidly and strongly induced the expression of both TaBOX and TaERF,with the mRNA levels of TaBOX and TaERF in shoots peaking at respectively 10 and 4 h and then decreasing(Fig.4A, B); in contrast, the mRNA levels of TaBOX and TaERF in roots peaked at respectively 4 and 10 h and the decreased(Fig.S13A,B).The expression of TaBOX was increased,whereas that of TaERF was reduced after LN treatment in shoots (Fig.4C, D)(two-sided t-test, P < 0.05).Varieties with high PDWR or CIR displayed stronger expression of TaBOX and TaERF in shoots in response to N stress than did those with low PDWR or CIR vales.This finding was consistent with the PDWR or CIR values (Fig.4E,F).The expression trend of TaBOX in roots was similar to that in shoots (Fig.S13C, E).In contrast, the expression of TaERF in the roots was not altered in response to N stress(Fig.S13D,F).To verify the function of the candidate gene TaBOX,we obtained one EMS mutant strain, carrying a termination mutation in the exon of TaBOX, from the Wheat Jing 411 TILLING Database (Fig.5A, B).We compared the phenotypes of the Tabox mutant and wild-type plants (WT) under HN and LN conditions.The Tabox mutant showed significantly lower RL, SFW, SDW, RFW, RDW, PFW, and PDW than the WT under LN conditions(Figs.5C,S14).These differences suggested that TaBOX might be the causal gene for qPDWR4B.1 and TaERF the causal gene for qCIR1A.3.

Fig.3.Variation in qCIR1A.3 haplotypes controlling chlorate inhibition rate.(A) Local Manhattan plot and linkage disequilibrium (LD) heat map of single-nucleotide polymorphisms(SNPs)within the LD block of qCIR1A.3.CIR,chlorate inhibition rate.(B)Three haplotypes(Hap 1,Hap 2,and Hap 3)were identified for qCIR1A.3 based on the variations in 15 SNPs.Hap,haplotype.(C)Haplotype analysis of qCIR1A.3.Violin plot indicates the distribution of CIR in each haplotype.Different letters represent significant differences at P < 0.05.

Fig.4.Expression of two candidate genes in NUE-associated QTL.(A)Expression levels of TaBOX(A)and TaERF(B)after nitrate treatment in shoots.Different letters indicate differences(P= 0.05);error bars indicate ±SD;values are mean± SD of three replicates.(C)Expression of TaBOX in the shoots of five Low-PDWR and High-PDWR varieties.PDWR, N sensitive index of PDW.(D) Expression of TaERF in the shoots of five Low-CIR and High-CIR varieties.CIR, chlorate inhibition rate.Values are mean ± SD of three independent samples.Relative expression levels of target genes were normalised to the expression of TaACTIN.Expression response(change in expression level relative to an initial low level of expression under various N conditions)of TaBOX(E)and TaERF(F)to N stress(ERN)in shoots and corresponding PDWR or CIR values.For ERN,values are mean±SD of three biologically independent samples.For PDWR and CIR,n=10;wheat plants were grown for 14 d in nutrient solutions containing 2 mmol L-1(HN)or 0.2 mmol L-1(LN)nitrate.The relative expression levels of target genes were normalized to the expression of TaACTIN.PDW,plant dry weight;PDWR,N sensitivity index of PDW
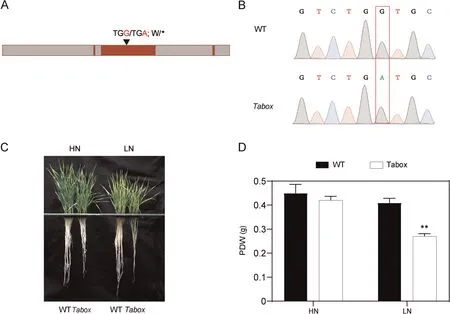
Fig.5.Phenotypic differences between the Tabox EMS mutant and wild type(Jing 411)plants under high(HN)and low(LN)N conditions.(A)Gene structure of Tabox with the mutant site.(B)Sequence chromatograms of TaBOX in the wild type and EMS mutant covering the mutation site.(C)Phenotypes of Tabox mutant and wild type wheat plants under LN and HN conditions for 3 weeks.(D)Phenotypic difference in PDW between the Tabox EMS mutant and wild type Jing 411 under HN and LN conditions.Means were compared with a t-test.** indicates significance at P < 0.01.PDW, plant dry weight.
4.Discussion
4.1.Identification of QTL and candidate genes associated with NUE in wheat seedlings using GWAS
Comparing agronomic efficiency using LN and HN inputs is an effective approach for evaluating NUE[38].Various agronomic efficiency indicators, including aboveground, whole plant, and root biomass, aboveground N accumulation, total N absorption of plants,SPAD value,leaf area,and PH,are used for the NUE evaluation of crops[39–41].Qin et al.[42]showed that under controlled N treatment conditions, the fresh weight and N accumulation of the whole plant of LN-sensitive and LN-tolerant germplasm were higher than those after LN treatment.They also found that under conditions of controlled N supply, the correlation between fresh weight per plant and dry weight above ground was highest.In the present study,all 13 wheat seedling traits investigated,except RL and RSR, were increased under HN treatment compared with those after LN treatment; in contrast, SFW was reduced by 40.22% (Figs.S1–S3).PH was negatively correlated with all traits but RL and LL under HN conditions; RFW, SFW, RDW, SDW, PFW,and PDW showed significant correlations (Fig.S4A).The correlations among agronomic traits under LN conditions were similar to those under HN conditions (Fig.S4B).The co-location of some traits was consistent with the results of correlation analysis;RDW and RFW were mainly co-located on chromosome 2A,whereas LL and LW were clustered on chromosome 3A (Table S4).
Recently,GWAS has led to the advancement of genetic research related to wheat NUE.A GWAS of five root architecture-related traits in wheat was performed using a 660K SNP array at the seeding stage under conditions of different N levels[43].A nested association mapping (NAM) population containing 2059 SNP markers from 90K SNP chips was used for a GWAS of N use in wheat seedlings[44].A GWAS of 14 traits associated with biomass and NUE in 134 wheat varieties at the seedling stage was perfored using a 90K SNP array [45], resulting in the identification of five SNPs for six traits and two SNPs for multiple environments.In the present study, we conducted a GWAS using 397,384 SNPs for 13 NUEassociated traits at the seedling stage (Table S2) and detected 49 QTL for NUE-related traits (Table S4).
Of the 135 candidate genes screened from all annotated genes(Table S6), 71 were upregulated or downregulated after LN treatment.We found several candidate genes associated with N utilisation;these included TraesCS6A02G017500 encoding a nitrate reductase, TraesCS1A02G291100 encoding an aminotransferaserelated family protein, TraesCS4B02G385400 encoding a glycosyltransferase,TraesCS4A02G439000 encoding an oligopeptide transporter, and TraesCS1A02G330400 encoding a phosphate transporter.We also identified several genes possibly involved in senescence-associated development, including TraesCS1A02G350900 encoding a senescence regulator, TraesCS4A02G439000 encoding an oligopeptide transporter, and TraesCS1A02G329400 encoding a serine/threonine-protein kinase.As the N uptake of wheat continues up to the leaf senescence stage,delayed senescence leads to an increase in NUE by allowing the plant to take up more N [46].This trait is thought [47] to be associated with increased leaf N levels, resulting in high N and carbon allocation for root growth.
In the present study, qPDWR4B.1 was identified as one of eight stable QTL, with TaBOX being identified as its candidate gene(Fig.2).This gene was annotated as an F-box family protein and displayed a strong expression in response to N deficiency.This Fbox protein gene is reportedly involved in the expression of various phytohormones during abiotic stresses [48,49].The finding that the Tabox mutant showed lower PDW than the WT under LN conditions indicates that TaBOX positively regulates NUE under LN conditions.
We identified Hap 1 as the favorable allele of TaBOX for a stable PDWR in response to LN stress (Fig.2C).The frequency of a favorable allele in the public dataset was 0.19(Fig.S11).Although Hap 1 appeared to be one of the prominent haplotypes, this finding also suggested the absence of favorable haplotypes in numerous modern cultivated varieties.
4.2.Chlorate sensitivity test is a useful tool for determining the NUE in wheat
Because chlorate is a nitrate analog that uses the same absorption and assimilation pathways as does nitrate,the chlorate sensitivity test can be used for phenotypic evaluation by simulating nitrate absorption by plants.To our knowledge, this is the first study to conduct a GWAS using chlorate resistance to identify NUE-associated QTL in wheat at the seedling stage.After chlorate treatment, the highest CIR of each variety in this population was 53.93% (Table S3), the lowest was 2.15%, and the mean was 28.77%.In the GWAS of CIR, we identified six QTL related to CIR;qCIR1A.5 overlapped with the reported QTL QRDW-1A.1 [50],qCIR5B.1 overlapped with gwm371, and qCIR1A.3 overlapped with wsnp_Ra_c4664_8410628 [51] (Table S4).We speculated that TaERF, which displayed a strong expression in response to N deficiency in shoots, was the causal gene for qCIR1A.3 (Fig.3B).However, the expression of TaERF in roots did not change in response to N deficiency(Fig.S13 D,F),suggesting the presence of a mobile signal from the root to the shoot for modulating NUE.Because its homolog in Arabidopsis, AT3G23240, is reportedly [37] involved in nitrate assimilation,we speculate that TaERF functions in crosstalk between N and ethylene in wheat.
4.3.Co-localization of the seedling and maturity stages
Compared with field experiments, seedling experiments offer relatively controllable environmental conditions and facilitate the collection of more precise phenotypic data, providing a crucial foundation for obtaining reliable SNP association loci.In our previous study, we detected 11 stable QTL for NUE-associated agronomic traits out of 347 QTL at the maturity stage [18].In the present study,we detected eight stable QTL of 49 QTL at the seedling stage (Table S4).Ten QTL detected at the seedling stage overlapped with the 8 NUE-related agronomic traits detected at the maturity stage in our previous study.These overlaps included qLLR3A.2 with qPHR3A.2, qPFWR5B.1 with qTKWR5B.2, qRFWR1A.1 with qTNR1A.3, qRFWR2B.1 with qBpPR2B.2, qRLR2B.1 with qGYPR2B.3, qRSRR1D.1 with qPHR1D.1, qRSRR2B.1 with qGNSR2B.1,qSFWR5B.1 with qTKWR5B.2,qCIR1A.4 with qFsnR1A.2,and qCIR1A.5 with qTNR1A.2.TraesCS2B02G368500 is a strong candidate gene for the QTL qRSRR2B.1 and qGNSR2B.1, as it encodes a nitrate transporter 1.2, which is involved in low-affinity nitrate uptake, and was downregulated following LN treatment.These QTL may regulate NUE at both the seedling and maturity stages.
5.Conclusions
Of 49 QTL associated with NUE in a 389-accession wheat panel,eight were identified as multi-environment stable QTL, 27 overlapped or were close to reported QTL, and 22 may be novel QTL.Two genes TaBOX and TaERF, showed strong expression responses to N, making them strong candidates for genes controlling NUE.A Tabox mutant showed more sensitivity to LN stress than the wild type.Hap 1 is the favorable allele of TaBOX for stable PDWR in response to low N stress.Two functional markers for TaBOX were developed.
CRediT authorship contribution statement
Huawei Shi:Conceptualization, Formal analysis, Investigation.Weichong Wang:Conceptualization, Formal analysis, Investigation.Lifeng Gao:Investigation, Validation, Visualization.Jirong Wu:Investigation, Validation.Chengmei Hu:Investigation.Huishu Yan:Visualization, Formal analysis.Yugang Shi:Validation.Ning Li:Validation.Youzhi Ma:Resources.Yongbin Zhou:Validation.Zhaoshi Xu:Resources.Jun Chen:Project administration.Wensi Tang:Resources.Kai Chen:Resources.Daizhen Sun:Conceptualization, Supervision, Project administration, Funding acquisition.Yuxiang Wu:Writing–review& editing,Supervision,Project administration,Funding acquisition.Ming Chen:Conceptualization,Writing–review&editing,Supervision,Project administration, Funding acquisition.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This work was supported by the National Key Research and Development Program of China(2022YFD1200201),Henan Provincial Science and Technology Research and Development Plan Joint Fund (222301420025), and the Agricultural Science and Technology Innovation Program (ASTIP) of CAAS.The authors would like to thank professor Jizeng Jia for providing this study’s natural materials.
Appendix A.Supplementary data
Supplementary data for this article can be found online at https://doi.org/10.1016/j.cj.2023.10.014.

- The Crop Journal的其它文章
- Flowering-time regulation by the circadian clock: From Arabidopsis to crops
- Global characterization of OsPIP aquaporins reveals that the H2O2 transporter OsPIP2;6 increases resistance to rice blast
- Drought-triggered repression of miR166 promotes drought tolerance in soybean
- The OsBSK1-2-MAPK module regulates blast resistance in rice
- Natural variation of an autophagy-family gene among rice subspecies affects grain size and weight
- Rice gene OsUGT75A regulates seedling emergence under deep-sowing conditions