
Interfacial reinforcement of core-shell HMX@energetic polymer composites featuring enhanced thermal and safety performance
2024-02-29 08:23:18BinghuiDunHongchngMoBojunTnXinmingLuBozhouWngNingLiu
Defence Technology 2024年1期
Binghui Dun , Hongchng Mo , Bojun Tn , Xinming Lu , Bozhou Wng ,b,*,Ning Liu ,b,**
a Xi'an Modern Chemistry Research Institute, Xi'an, 710065, People's Republic of China
b State Key Laboratory of Fluorine & Nitrogen Chemicals, Xi'an, 710065, People's Republic of China
Keywords: HMX crystals Polyalcohol bonding agent Energetic polymer Core-shell structure Interfacial reinforcement
ABSTRACT The weak interface interaction and solid-solid phase transition have long been a conundrum for 1,3,5,7-tetranitro-1,3,5,7-tetraazacyclooctane (HMX)-based polymer-bonded explosives (PBX).A two-step strategy that involves the pretreatment of HMX to endow -OH groups on the surface via polyalcohol bonding agent modification and in situ coating with nitrate ester-containing polymer, was proposed to address the problem.Two types of energetic polyether-glycidyl azide polymer (GAP) and nitrate modified GAP (GNP) were grafted onto HMX crystal based on isocyanate addition reaction bridged through neutral polymeric bonding agent(NPBA)layer.The morphology and structure of the HMX-based composites were characterized in detail and the core-shell structure was validated.The grafted polymers obviously enhanced the adhesion force between HMX crystals and fluoropolymer (F2314) binder.Due to the interfacial reinforcement among the components, the two HMX-based composites exhibited a remarkable increment of phase transition peak temperature by 10.2°C and 19.6°C with no more than 1.5%shell content, respectively.Furthermore, the impact and friction sensitivity of the composites decreased significantly as a result of the barrier produced by the grafted polymers.These findings will enhance the future prospects for the interface design of energetic composites aiming to solve the weak interface and safety concerns.
1.Introduction
Polymer-bonded explosives (PBX), in which explosive particles are held together by polymer binders,are widely applied in current defense and civilian field[1-5].After the formulation is confirmed,the interface structure and interactions between different components largely determine the quality and performance of PBX.The worry is that the interfacial interaction between polymer binder and energetic crystals is so weak that the fracture and crack along explosive-binder interface is the most common problem in the application of PBX[6-10].Therefore,designing novel and efficient interfacial structure to enhance the interfacial interaction is highly urgent in this field.Besides, the safety concerns brought by the pursuit of high energy need to be paid sufficient attention in consideration of handling, transport and storage of PBX.Extensive research efforts have been devoted in terms of desensitization of energetic materials (EMs), such as recrystallization, refining, cocrystallization and surface coating[11-17].Therein, surface coating stands out with tunable and integrated performance without damaging the chemical structures of the pure explosives.
There are several techniques of surface coating for EMs,such as water suspension method,emulsion method,spray drying method and crystallization coating method [18-22].However, these coating methods suffer from the weak interfacial adhesion force and considerable amount of additives needed, resulting in low surface coverage and limited coating efficiency.The EMs with low sensitivity and high energy witnessed the development ofinsitucoating in the last decade.As a direct chemical coating process,in situcoating exhibits high efficiency with few reactants acquired to achieve perfect coating and profitable processing performance[23-25].For instance, Yang et al.have reported nitramine explosives@melamine-formaldehyde (MF) resins composites with markedly decreased sensitivity [26].Bio-inspired coating of polydopamine (PDA) on 1,3,5,7-tetranitro-1,3,5,7-tetraazacyclooctane(HMX) gained well-coated composites with enhanced thermal stability [27].However, the high cost of raw materials, alkaline coating atmosphere and poor desensitization effect due to the rigid nature of PDA shell restrict the wide applications of PDA in PBX formulations.Withinsitucoating of PDA on 1,3,5-triamino-2,4,6-trinitrobenzene (TATB) particles and further grafting hyperbranched polymers on PDA surface, the core@double shell composites feature improved mechanical properties [28].In principle,insitustrategy could be an effective route to tune the performance of explosives for specific needs [29-32].
As one of the most attractive base explosive in PBX formulations, HMX has a high detonation velocity and pressure [33].This draws a considerable attention due to its potential application in defense area where the pursuit of high energy is always the target.However, there are still various technical problems waiting for solutions before a broad application of HMX.One is that the mechanically brittle characteristic and relatively high sensitivity of HMX crystals improve the difficulty of process handling[34,35],the other is that HMX undergoes phase transition from the stable β form to sensitive δ form when exposed to thermal insult [36,37].Therefore, seeking effective techniques to control the phase transition and decrease the sensitivity of HMX is highly necessary.Glycidyl azide polymer (GAP), featuring natural -OH and -N3 groups on its molecule, has been widely applied to improve the stability and energy releasing performance of aluminum particles by constructing core-shell composites [14,38].The GAP polymers could serve as energetic shell and enhance the adhesion force with polymeric binders.However, the GAP coating suffers from poor interchain interaction and weak mechanical properties due to the existence of -N3group.
Herein, a novel polyether polymer-1,2,3-triazole-4-methylene nitrate modified azide polymer (GNP) was first synthesized via Huisgen cycloaddition reaction to introduce nitrate ester group in side chain of GAP.In order to obtain high coverage and adhesion force,HMX crystals were pretreated by neutral polymeric bonding agent (NPBA) to impart -OH groups on the surface, which can be used as reaction sites for further grafting to achieve the purpose of functional regulation.The structure of HMX and NPBA were presented in Fig.1.Two grafted polymers(GAP and GNP)were adopted to modify HMX to achieve enhanced interfacial and safety performance via in situ coating.The morphology and structure of the core-shell composites would be first investigated, and then the effects of surface coating on the surface properties, thermal behavior and sensitivity of the composites were explored in detail.With the enhanced thermal and safety performance,HMX-N@GAP and HMX-N@GNP composites are expected to replace HMX in PBXs to address the safety concerns.The obtained composites could be applied to pressed PBX with fluoropolymer as binder or cast-cured PBX formulations with polyurethane as binder.
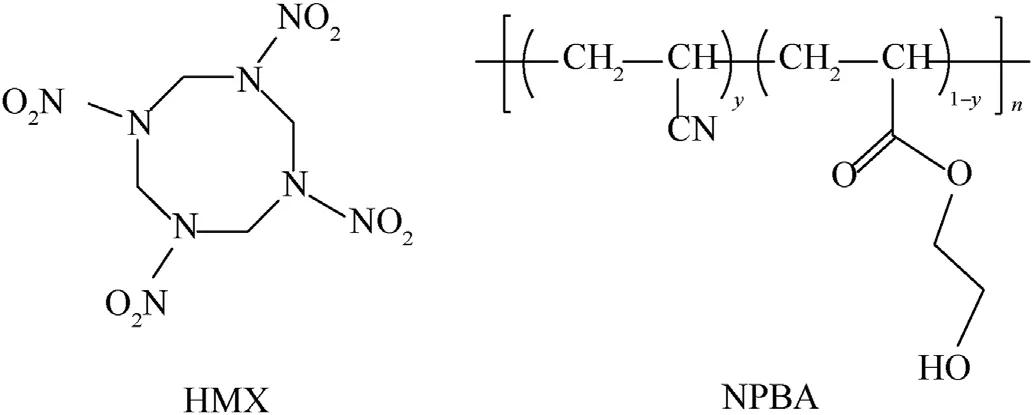
Fig.1.Chemical structures of HMX and NPBA.
2.Experimental
2.1.Materials
HMX was bought from Yinguang Chemical Industry Group Co.,Ltd.(Gansu, China).Glycidyl azide polymer (GAP), propargyl alcohol nitrate,polyaryl polymethylene isocyanate(PAPI,99%)and neutral polymeric bonding agent(NPBA)were obtained from Xi'an Modern Chemistry Research Institute (Shannxi, China).The molecular mass (Mn) of GAP was 4000 g/mol, with the polydispersity index (Mw/Mn) of 1.6.Dibutyltin dilaurate (DBTDL, 95% solution),1,2-dichloroethane acting as solvent were purchased from Aladdin(Shanghai, China).
2.2.Sample preparation
Preparation of GNP.5 g GAP was completely dissolved in 25 mL 1,2-dichloroethane under vigorous magnetic stirring for 5 min.Then, 0.5 g propargyl alcohol nitrate was added dropwise.The solution was placed in a water bath at 65°C for 5 h.After removing the solvent by reduced pressure distillation, the yellow oil was obtained finally.After drying, 4.6 g GNP was weighed.
Modification of HMX via NPBA.5 g HMX was dispersed in 50 mL 1,2-dichloroethane through high-intensity ultrasonic irradiation in an ultrasonic apparatus for 20 min.Then,0.025 g NPBA was added to the solution and maintained ultrasonic treatment for 2 h.Finally,the modified HMX particles(marked as HMX-N)were obtained by vacuum filtration and dried at 60°C for 24 h.
Coating of HMX-N with interface-reinforced polymers.2 g HMXN was put into a three-necked, round-bottomed flask with 40 mL 1,2-dichloroethane.0.06 g grafted polymers(GAP,GNP),0.08 g PAPI dissolved in 1,2-dichloroethane were added under vigorous stirring successively.The solution was heated to 35°C and 0.8 mg DBTDL dissolved in 1,2-dichloroethane (4 mg/mL) was injected into the suspension.After several hours, the polymer grafting was completed.The target composites were filtered, washed with 1,2-dichloroethane and dried at 60°C for 24 h.
2.3.Characterization
The structure of GNP was examined by infrared spectra(IR),1H nuclear magnetic resonance (NMR),13C NMR and gel permeation chromatography(GPC).The raw HMX and coated-HMX composites were detected through scanning electron microscope (SEM),atomic force microscope (AFM), powder X-ray diffraction (PXRD),X-ray photoelectron spectroscopy(XPS),Raman spectra and IR.The component content of explosive was determined by highperformance liquid chromatography (HPLC) analysis based on an external standard method.The surface, thermal properties and sensitivity of the composites were measured by contact-angle test,TG-DSC, the characteristic height (H50) and explosion probability(P), respectively.The detailed characterization methods were presented in the Supporting Information.
3.Results and discussion
3.1.Preparation and characterization of GNP
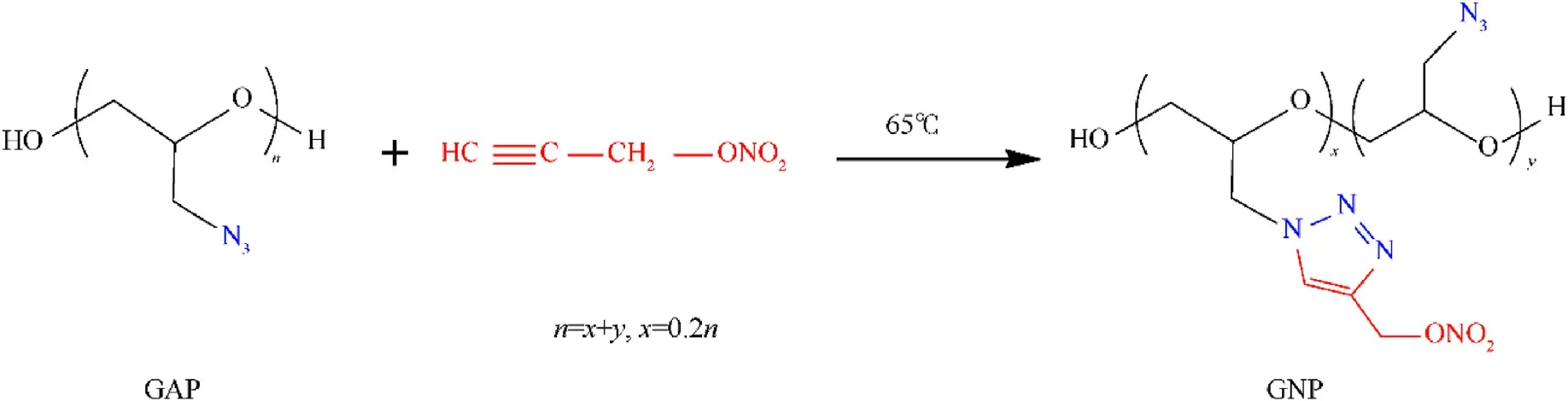
Scheme 1.Synthetic route of GNP.
An amount of GNP was preparedviaHuisgen cycloaddition reaction using GAP and propargyl alcohol nitrate as raw materials,as shown in Scheme 1.The chemical structure of GNP was first investigated by IR tests,and the IR spectrum of GAP was studied as well in comparison (Fig.2).A strong peak around 2100 cm-1,denoting the typical absorption of‒N3group,was observed for GAP and GNP.As compared to GAP, the additional peaks at 1726 cm-1and 1626 cm-1correspond to the stretching vibration of C=C and-ONO2group.As the percent of substituted -ONO2group in GNP molecule is only 20%,the absorption peak of-ONO2in IR spectrum is weak.The chemical structure of GNP was also characterized by1H NMR, as shown in Fig.S1.The peaks at 3.35-3.66 ppm and 3.73 ppm are attributed to methylene protons and methine protons connected to ether groups, respectively.The peak at 5.61 ppm is ascribed to the hydrogen protons on the triazole ring.Therefore,it could be confirmed that GNP has been successfully prepared.The GPC measurement reveals that the molecular mass(Mn)of GAP was 5468 g/mol, with the polydispersity index of 1.53.
3.2.The proposed formation mechanism of HMX-based core-shell composites

Fig.2.IR spectra of GAP and GNP.
The preparation process and proposed formation mechanism of HMX-based core-shell composites are displayed in Scheme 2.The preparation process involves two steps,namely NPBA modification on HMX crystal andinsitucoating of modified HMX.At first,NPBA modification confers -OH group on the surface of HMX under ultrasonic irradiation.Driven by hydrogen bonding interactions between HMX and NPBA, where -OH groups in the NPBA acted as proton donors and -NO2groups in the HMX as proton acceptors,the grain NPNA molecules accumulated on the surface of HMX uniformly.This step plays a vital role for the subsequent in situ deposition of the polymers,where NPBA layer serves as a bridge to give anchors for polymer grafting.Then, two kinds of polymers were grafted onto HMX-N via addition reaction between-OH and isocyanate lipid groups (-NCO).-NCO groups of PAPI at the interval site would react separately with the active -OH groups of NPBA and the selective polymers under specific conditions.The cross-linking network structure was constructed across the interface of HMX-N with the in-depth addition reaction and molecular chain extension.The resultant shell can protect the HMX crystals from deformation which is desirable for the suppression of phase transition, and behave as a buffer layer to dissipate the impact energy.
3.3.Morphology
The effectiveness of surface coating is commonly checked by examining the morphology changes, especially the variation of surface roughness.The morphologies of HMX crystal particles were measured through SEM images and displayed in Fig.3.It could be found from Fig.3(a) that raw HMX crystals exhibit polygonal morphology with an average particle size of 23 μm.After NPBA modification, HMX crystals were covered by a layer of granular particles (Fig.3(b)).One should note that cracks appeared on the surface of HMX-N under the electron beam during high-resolution SEM test.The sensitive nature of energetic crystals to electron beam was significantly improved in the well-coated samples due to the dense protective shell formed by GAP and GNP polymers.The shell content in the composites was controlled by tuning the reaction time and calculated by formula 100% - [content of energetic crystals].The content of HMX could be obtained by core-etching test and HPLC measurement.The methods were detailed in the Supporting Information.The GAP/GNP contents for HMX-N@GAP-2h, HMX-N@GAP-4h, HMX-N@GAP-6h, HMX-N@GNP-2h, HMXN@GNP-4h and HMX-N@GNP-6h composites are 0.7%, 1.3%, 1.8%,0.8%,1.5%and 2.0%,respectively.One can observe that the surface of HMX-N was completely and compactly coated viainsitudeposition.The HMX composites appear light agglomeration, which is due to the strong cohesion of GAP and GNP polymers.With the increasing reaction time, the content and thickness of the polymer shell increased accordingly.In consideration of coating efficiency and energy loss, theinsitureaction time of 4 h is thought to be appropriate.In comparison, HMX/GNP composite was prepared with the reaction time of 4 h.It is obvious that the polymers could not encapsulate HMX core perfectly with considerable bare parts and agglomerations observed (Fig.3(i)).It suggested that the modification of HMX crystals with polyalcohol NPBA can impede the self-nucleation of polymers and afford more growing sites to improve the coating coverage.
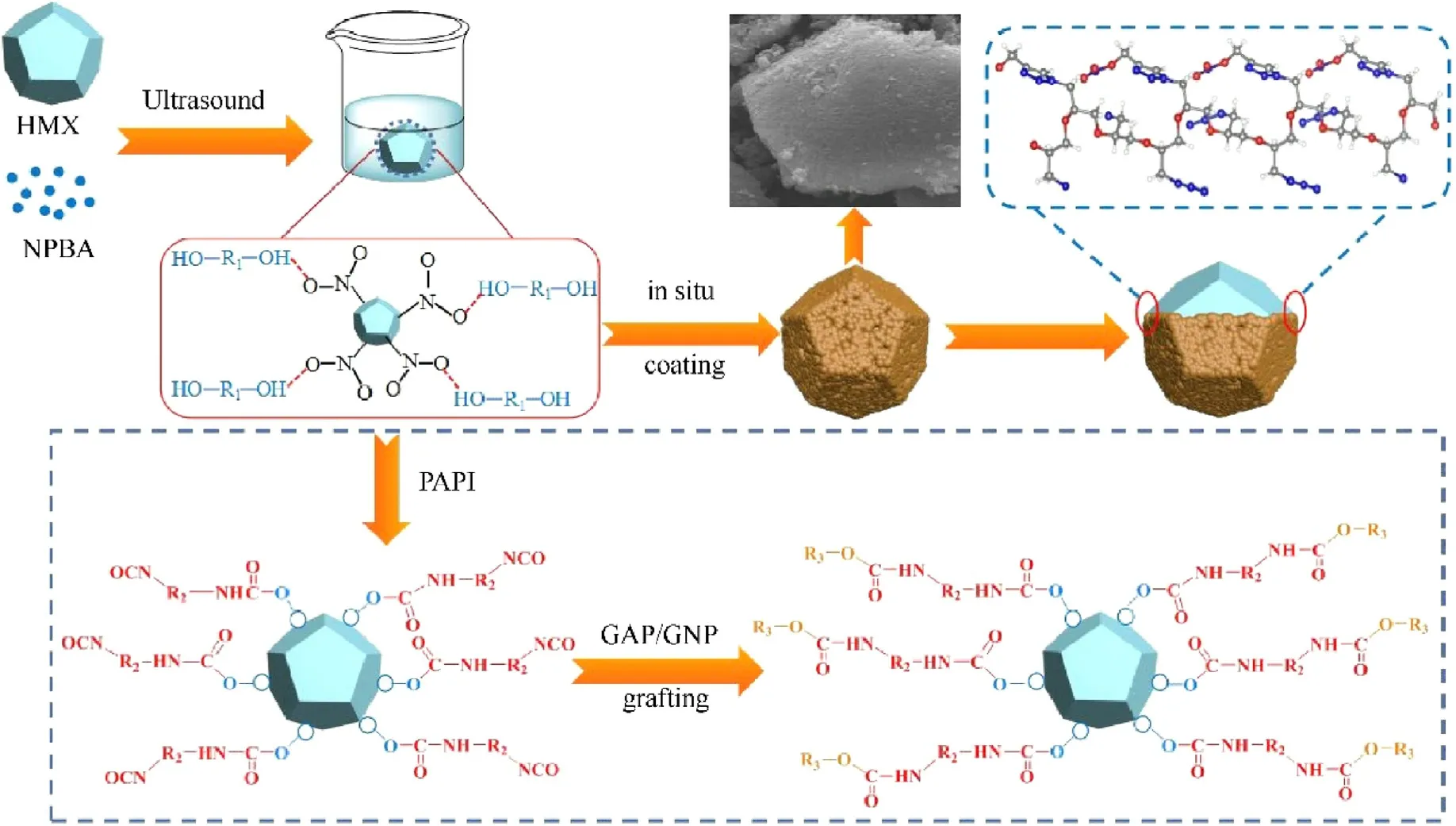
Scheme 2.The preparation process and proposed formation mechanism of HMX-based core-shell composites.
AFM measurements were further carried out to evaluate the surface roughness and morphology of raw HMX and the coated HMX particles, and the AFM images were demonstrated in Fig.4.Table 1 gave the surface roughness parameters of the samples.It can be found from Fig.4(a) that the raw HMX crystals exhibit a slight undulating surface.After NPBA modification, the surface becomes relatively uniform withRa= 8.1 nm andRq= 19.1 nm(Fig.4(b)).The adhesion of NPBA on HMX crystal produces considerableinsitureaction points and increases the surface roughness evidently.The variation of surface morphology along withinsitureaction time was explored in Figs.4(c)-4(h).When the reaction time was short(2 h),the surface roughness decreases and the coated polymers arrange regular.This phenomenon was attributed to that a more homogeneous surface deposition might be achieved due to the conjugation of GAP or GNP aggregates.With the increase of reaction time, the surface roughness continues to increase, together with a compact and intact surface morphology.This also suggests that GAP and GNP have successfully deposited on the HMX-N surface and the polymers tend to form firm, enduring shells onto the HMX surfaces instead of self-agglomeration.Moreover, the surface roughness of GNP-coated particles is larger than that of GAP-coated ones and a more even surface was found for GNP-coated composites.It is deemed that the coating effect is more desirable with GNP as coating shell and the interface interaction between GNP shell and HMX core may be stronger.
3.4.Structure characterizations
The core-shell structure of HMX-N@GAP and HMX-N@GNP composites were studied by XRD, Raman and IR measurements and the corresponding spectra were presented in Fig.5.HMXN@GAP and HMX-N@GNP composites withinsitureaction time of 4 h were adopted as examples.It can be found from Fig.5(a)that HMX exhibited obvious β-crystalline structure with the characteristic diffraction peaks(14.80°,16.02°,20.51°,23.03°,29.65°,31.90°).As we can see that the peak position and pattern of the HMX-based composites were similar to HMX, suggesting that no crystal transformation occurred during theinsitureaction process.It is worthy to note that the addition of GAP or GNP influences some peak intensities of HMX crystals.As an illustration,the intensity of peaks at 14.80°, 20.51°decreases when compared to pure crystal,while 2θ at 31.90°increases for HMX-N@GAP composite.When it comes to HMX-N@GNP composite, the peak intensity of 2θ at 16.02°,20.51°increases in comparison with raw HMX crystal,while 2θ at 29.65°decreases.It can be traceable to the fact that coating of GAP or GNP induces preferred orientation of HMX crystals.
Raman is an effective route to identify the chemical characteristics of materials without destroying their structures.One can observe from Fig.5(b)that all the diffraction peaks of HMX can be classified as β-HMX.The results of Raman spectra come to the same conclusion with XRD study that the polymorph of HMX does not change in the whole coating process.The peaks between 1100 cm-1and 1500 cm-1are attributed to the stretching vibration of -NO2for pure HMX.Afterinsitucoating, the intensity of peaks in this region enhanced, especially for HMX-N@GNP composite.The reason may be that the stretching vibration of the double bonds,such as C=C,N=N and C=O in GAP and GNP structures lies in this section as well and the overlap of these peaks reinforced the absorption intensity.One should note that the intensity of peaks in 800-1000 cm-1, which belong to ring stretching, varies after modification.
Fig.5(c) reveals the IR spectra of HMX and the HMX-based composites.The characteristic stretching vibration of C-H bond and-NO2group of HMX could be observed around 3006 cm-1and 1560 cm-1.The IR spectra of HMX-N and raw HMX are almost the same due to the extremely low content of NPBA.After GAP or GNP coating, the featured diffraction signals of HMX crystal were maintained.The region in 2500-2000 cm-1was partially enlarged,as shown in Fig.5(d).It is clear that the characteristic peak of-N3group at 2103 cm-1appears in HMX-N@GAP and HMX-N@GNP composite, indicating that the HMX crystals are successfully encapsulated by low contents of GAP or GNP polymers.

Fig.3.SEM images of(a)raw HMX,(b)HMX-N,(c)HMX-N@GAP-2h,(d)HMX-N@GAP-4h,(e)HMX-N@GAP-6h,(f)HMX-N@GNP-2h,(g)HMX-N@GNP-4h,(h)HMX-N@GNP-6h and(i) HMX@GNP-4h composite.
The element composition and state of the composites surface were analyzed by XPS measurements,and the C 1s,N 1s,and O 1s spectra are depicted in Fig.6.Table 2 presents the surface element compositions of pure HMX and the HMX-based composites.For raw HMX,two peaks centered at 287.8 eV and 284.8 eV were observed in the C 1s spectrum,corresponding to N-C-N and C-C(or C-H),respectively.The N 1s peaks are attributed to(407.0 eV)and(401.5 eV).The O 1s peak observed at 533.2 eV could be identified as.All the fitting results of HMX are in good agreement with the previous literatures [17,27,29,36,37].The C 1s,N 1s,and O 1s spectra of HMX-N exhibit the sum features of HMX and NPBA.The C 1s region is composed of two peaks,of which arerespectively.The N 1s spectrum possesses three peaks assigned toin sequence.The O 1s peaks are attributed to(532.9 eV) and(531.6 eV).One can find from C 1s and O 1s spectra that the intensity of the characteristic peaks of HMX decreases visibly.In addition,the carbon concentration and C/N ratio of HMX-N are higher than those of pure HMX (Table 2).These results prove the successful modification of NPBA on HMX surface.In terms of HMX-N@GAP composite, the C 1s profile is composed of three peaks,corresponding toat 288.7 eV,at 286.4 eV andat 284.8 eV, respectively.Two peaks indicative for(407.7 eV) and(401.1 eV)are fitted in the N 1s region.The peak at 533.5 eV fitted in O 1s region corresponds toIn the spectra of HMXN@GNP composite, the basic shapes of NPS spectra are similar to that of GAP-coated composite.Due to the existence of‒ONO2group in GNP molecule,the peak intensity ofin C 1s profile andin N 1s profile increased significantly in GNP-coated samples when compared to GAP-coated ones.The absorption peaks of -CN in C 1s and N 1s profiles disappeared,suggesting that HMX-N was fully covered in the coating process.The C/N ratio of the two composites increases evidently owing to the addition of the carbon-rich GAP and GNP polymers.Therein,GAP-coated composite possesses the highest carbon percent of 67.3% and C/N ratio of 5.0.
3.5.Surface analysis
Contact angle tests were conducted to evaluate the influence ofinsitucoating on the surface wettability of HMX crystals in different solutions and the interfacial interaction between energetic crystals and polymer binder in PBX matrix.The water contact angles of pure HMX, HMX-N, HMX-N@GAP-4h and HMX-N@GNP-4h composites are illustrated in Fig.7.The water contact angle of pure HMX was 66.5°.The low water wettability of HMX could be ascribed to its low surface energy and nonpolarity characteristics.After NPBA modification, the water contact angle of HMX-N decreased to 58.5°, as NPBA involves many hydrophilic groups like hydroxyl and cyano group.Then, byinsitudeposition of hydrophobic GAP/GNP shell on HMX-N surface, the water contact angles of the composites increase evidently.Therein, the improvement effect of HMX-N@GNP composite is more significant due to the existence of polar ‒ONO2group.

Fig.4.The AFM images of (a) raw HMX, (b) HMX-N, (c) HMX-N@GAP-2h, (d) HMX-N@GAP-4h, (e) HMX-N@GAP-6h, (f) HMX-N@GNP-2h, (g) HMX-N@GNP-4h and (h) HMXN@GNP-6h composite.

Table 1Surface roughness parameters of HMX crystals.
The surface energy(γ)and interfacial adhesion work(Wa)were determined from the contact angles of different droplets on the energetic crystals.The calculation methods are detailed in the Supporting Information.The surface energies of HMX, HMX-N,HMX-N@GAP-4h and HMX-N@GNP-4h composites, together with Wabetween HMX/the coated composites and fluoropolymer(F2314)were tabulated in Table 3.The surface energy componentsandof F2314are referenced to be 1.15 mN/m and 28.52 mN/m[39].It can be found from Table 3 that NPBA modification improves the surface energy of HMX crystals and promotes the adhesion between HMX crystals and F2314binder.After in situ coating, the resulted composites exhibit higher surface energy,consisting of the increased γdand decreased γp.As a result, the polar difference reduces, contributing to the interfacial dispersion according to the principle of polar similarity.The enhanced adhesion work comes to the conclusion that the interfacial interaction between HMX crystals and F2314binder enriches after surface coating, and the adhesion force between HMX-N@GNP composite and F2314is the highest.
In order to compare the interfacial adhesive intensity between HMX and GAP or GNP, DFT calculations were performed using the Vienna ab initio simulation package (VASP) [40,41].The computational methods were detailed in Supporting Information.The optimized composite structures are presented in Fig.8.The resulting energy curves are displayed in Fig.S2.The maximal potential barrier (εbreak) and maximal stress of the interface fraction(σbreak) were calculated by Ref.[42].

Fig.5.(a) XRD; (b) Raman; (c) IR and (d) partial enlarged IR spectra (2500-2000 cm-1) of raw HMX and the HMX-based composites.

Fig.6.XPS spectra of C 1s, N 1s, and O 1s regions for pure HMX, HMX-N and the HMX-based composites.

Table 2Surface element composition of HMX and the HMX-based composites.
The calculated adhesive energy (Ea), εbreakand σbreakvalues of HMX@GAP and HMX@GNP composites are listed in Table 4.As we know, the more negative the adhesive energy is, the stronger the adhesion force is.It can be observed that the adhesive energy of HMX@GNP is lower than that of HMX@GAP, indicating that GNP coating makes the notable effect on the interfacial interaction.Moreover, GNP-coated composite possesses higher interfacial intensity than GAP-coated one.The tight interfacial contact between HMX crystal and GNP may be explained from two aspects: one is that there exists hydrogen-bonding interaction, together with significant π-π interaction along the interface between the triazole ring of GNP and the N-heterocyclic ring of HMX.The rigid triazole ring contributes to the interfacial intensity as well.The other is that-ONO2and -NO2group participate in the relatively strong interfacial interaction.By contrast, the interface of GNP-coated composite can only be connected by weak hydrogen-bonding,resulting in poor interface interaction.The length of the polymer chain also affects the interface adhesion of the composite.
3.6.Thermal behavior

Fig.7.The water contact angles of (a) pure HMX, (b) HMX-N, (c) HMX-N@GAP-4h and (d) HMX-N@GNP-4h composite.
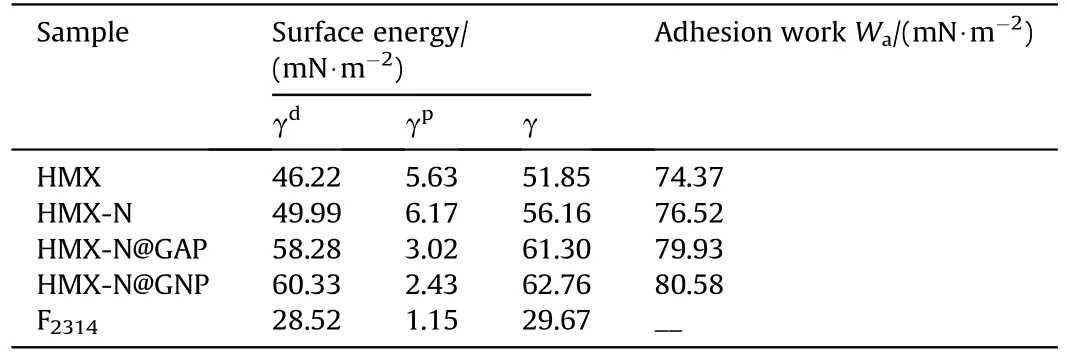
Table 3Surface energies of HMX, HMX-N, the HMX-based composites and F2314, together with Wa between HMX/the HMX-based composites and F2314.

Table 4The calculated adhesive energy (Ea), the maximal potential barrier (εbreak) and the maximal stress of the interface fraction (σbreak) of HMX@GAP and HMX@GNP composites.
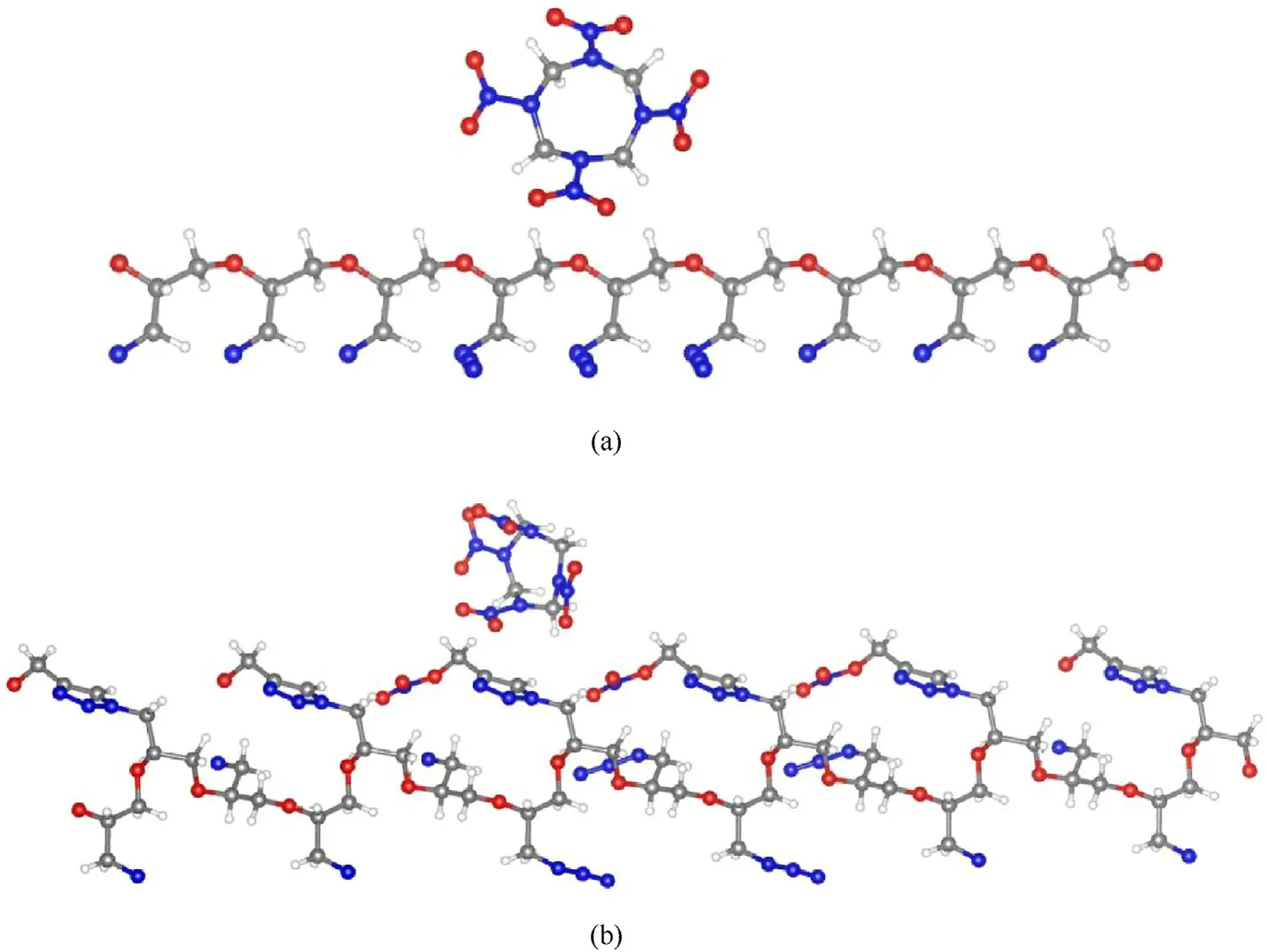
Fig.8.The optimized structures of (a) HMX@GAP and (b) HMX@GNP composite.
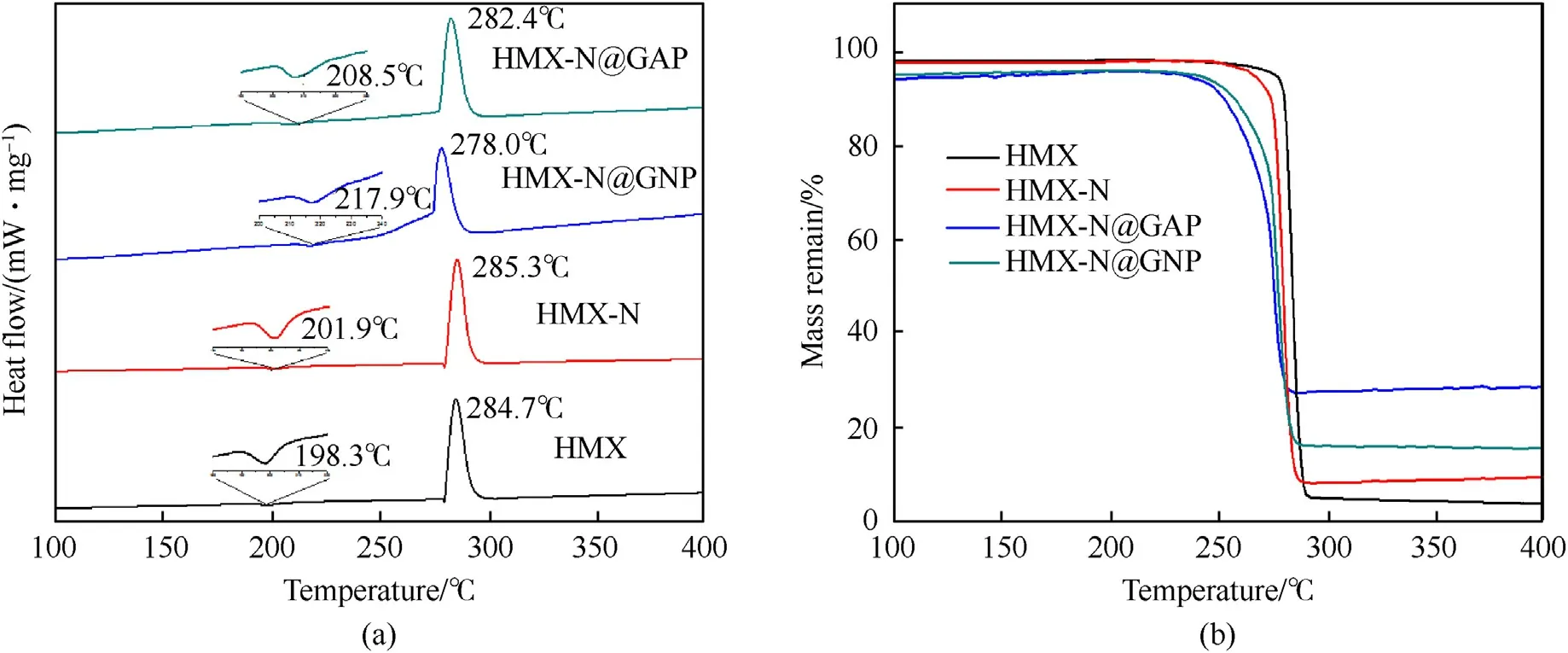
Fig.9.(a) DSC and (b) TG curves of HMX, HMX-N, HMX-N@GAP and HMX@GNP composites with the heating rate of 10 K/min.
To detect the thermal properties of HMX and the HMX-based composites, DSC and TG tests have been performed.The DSC and TG curves of pure HMX, HMX-N, HMX-N@GAP-4h and HMXN@GNP-4h composites with the heating rate of 10 K/min are presented in Fig.9.It can be observed from Fig.9(a)that HMX exhibits an endothermic peak at 198.3°C and a sharp exothermic peak at 284.7°C, which could be attributed to β→δ phase transition and thermal decomposition process in sequence.Compared with pure HMX, the phase transition and thermal decomposition peaks of HMX-N modified with NPBA moved to higher temperature slightly,signifying the positive effect of NPBA modification on the thermal stability of HMX.There is a remarkable increment of phase transition peak temperature by 10.2°C for HMX-N@GNP composite and 19.6°C for HMX-N@GAP composite, suggesting that the grafted polymers are good candidates for the enhanced polymorph stability of HMX through in situ coating.When it comes to thermal decomposition process, the peak temperature of HMX-N@GAP composite decreases compared with pure HMX.It may be attributed to the addition of GAP, which starts decomposition at 210°C and reaches the peak temperature at 253°C[14].As a contrast,GNP coating acts little on the thermal decomposition behavior.From TG curves of HMX and the coated-HMX composites (Fig.9(b)), it is clear that all the samples experience one-step weight lost,which is consistent with DSC study.The initial decomposition temperature decreases in the order of HMX >HMX-N >HMX-N@GNP >HMXN@GAP.
The kinetics of HMX and the coated-HMX composites were studied by non-isothermal DSC and the DSC curves with heating rate of 5,10,15 and 20 K/min were illustrated in Fig.10.It could be seen that all the four curves move to right as the heating rate increases.The thermal decomposition kinetic parameters of HMX and the coated-HMX composites are listed in Table 5.Take HMXN@GAP composite as example, the thermal decomposition peak temperature are 275.3, 277.3, 279.5 and 282.3°C in sequence.The thermal decomposition process can be assessed through Kissinger method in respect of activation energy (Ea) and pre-exponential factor (A).In general, lower activation energy suggests less energy required to reach the barrier of activation energy, resulting in a quicker induction of the decomposition reaction.TheEavalues for HMX-N@GAP and HMX-N@GNP composite obtained by Kissinger method are 329.1 kJ/mol and 341.7 kJ/mol, which are lower than those of single HMX,respectively.The results demonstrate that the thermal decomposition process is accelerated for the coated-HMX composites.The smaller values of A also convinced the enhanced thermal decomposition kinetics of the grafted HMX composites.
One should note that Kissinger method only takes the decomposition peak temperature in consideration without regarding the overall decomposition process, and sometimes error exists.Consequently,Friedman and combined kinetic methods have been adopted to calculate the kinetic parameters.TheEa-conversion rate(α) relationships for the thermal decomposition processes were plotted using Friedman method (Fig.S3).One can observe from Table 5 that all the correlation coefficients exceed 0.98, indicating that the kinetic evaluation results are reliable.As shown in Fig.S3,the Eavalues of HMX and the coated-HMX composites decrease all through the reaction process, implying that the thermal decomposition is autocatalytic as a result of the aggregation of active reaction sites.
The kinetic models for thermal decomposition process were expressed bymandnparameters in combined kinetic method.It can be seen from Table 5 that theEavalues obtained by Friedman method and combined kinetic method are approximate, implying that the calculation results are dependable.The obtained models of HMX and the coated-HMX composites were displayed in Fig.S4,and the ideal models were taken as reference.It is not hard to see that the decomposition reaction model of pure HMX is almost coincided with the curve of three-dimensional nucleation and nucleus growth model (A3).After NPBA modification, the thermal decomposition follows A3model in the initial period and moves towards two-dimensional nucleation and nucleus growth model(A2)in the range of α=0.56-0.99.With the further coating by GAP or GNP, the composites follow some model between A3and A2in the startup phase,then turn to random chain scission model(L2)in the later phase.One can speculate that the coated-HMX composites experience nucleation and nucleus growth at the beginning, and the chain scission degradation of crystal molecules is the intrinsical rate-limiting decomposition step of HMX-N@GAP and HMXN@GNP composites.
3.7.Sensitivity
The impact and friction sensitivity of HMX,HMX-N,the coated-HMX composites and the corresponding physical mixtures were studied, and the results are presented in Table 6.The impact sensitivity and friction sensitivity could be measured by characteristic height (H50) and explosion probability (P), respectively.HMX featuresH50of 26.0 cm andPof 98%.HMX-N shares the same level of sensitivity as HMX with a slight improvement of stability.It can be found that core-shell coating makes significant promotion on the mechanical stability of HMX crystals,especially compared to the physical mixtures.For instance, theH50of HMX-N@GNP-4h increases to 46.2 cm andPdecreases to 56%, while the physical mixture with the same component proportion makes unsatisfactory effect on the desensitization of HMX crystals.It may be attributed to the well-wrapped core-shell structure with HMX fully been covered, while physical mixing often results in partially bare surface and limited coating strength.The compact and intact GAP/GNP shell serves as a buffer to dissipate the abrupt impact or friction energy in the core-shell composites.With the reaction time goes on, the coverage and shell thickness increase accordingly,leading to more pronounced desensitization effect.Meanwhile,the sensitivity of HMX-N@GNP composites are lower than those of HMX-N@GAP ones.For one thing, GNP-coated particles feature more rough and uniform surface with few deformations or fractures, reducing the possibility of hot spots formation.For another thing, the interface interaction and intensity between GNP shell and HMX-N core are stronger owing to the existence of -ONO2group and triazole ring in GNP polymer.HMX-N@GNP-6h possesses the lowest sensitivity among the samples with H50of 53.4 cm and P of 42%.
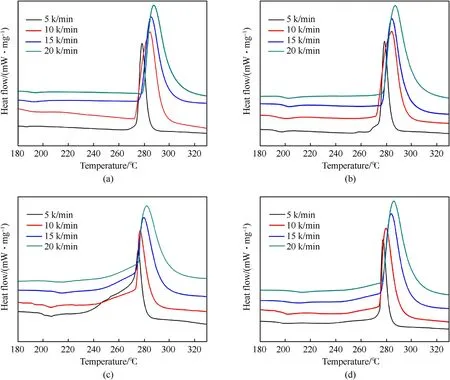
Fig.10.DSC curves at different heating rates for (a) HMX, (b) HMX-N, (c) HMX-N@GAP and (d) HMX-N@GNP composite.
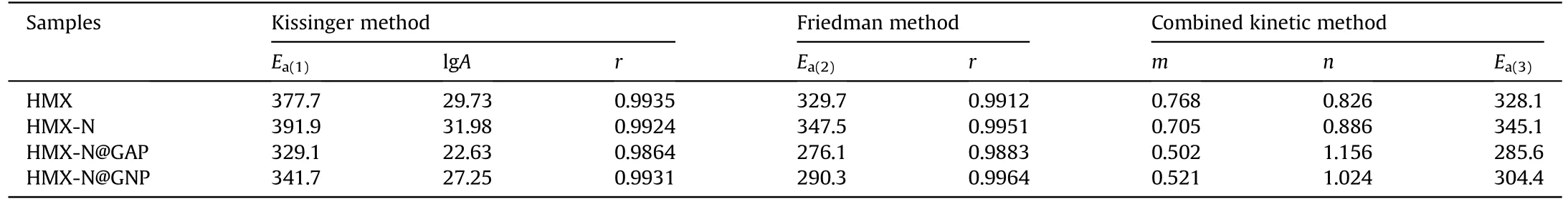
Table 5The thermal decomposition kinetic parameters of HMX and the coated-HMX composites.
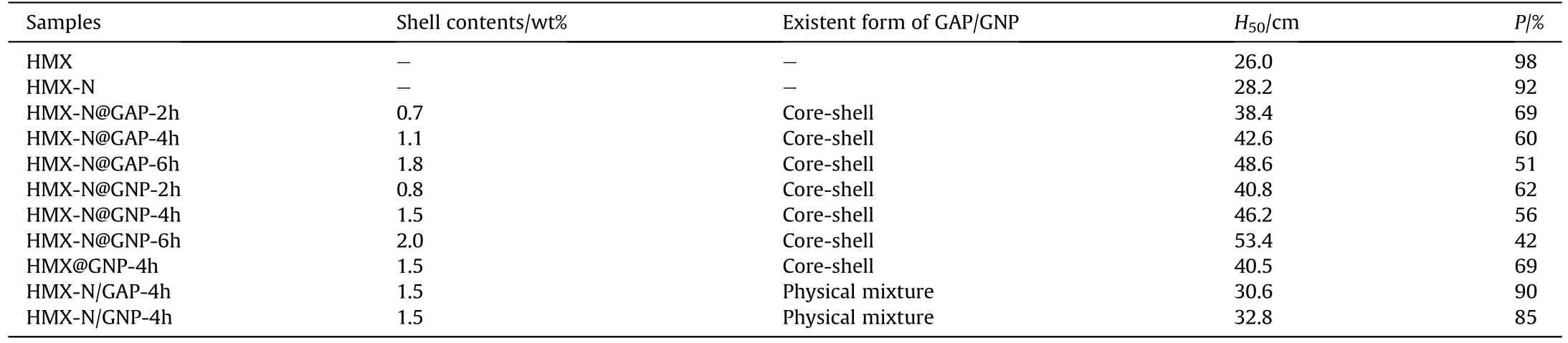
Table 6The impact and friction sensitivity of samples.
4.Conclusions
In this paper, NPBA with strong adhesion force was firstly used for HMX modification, and two types of polyether polymers (GAP and GNP) were grafted subsequently via addition reaction.A compact and intact core-shell structure of the composites was confirmed by comprehensive morphology and structure characterization.Theinsitureaction time of 4 h is desirable in consideration of coating efficiency and energy loss.From the results of contact angle tests, the hydrophobic GAP/GNP shell improved the water contact angle and enhanced the interfacial adhesion between HMX crystals and F2314binder.Besides, the polymorph stability of HMX was distinctly improved with the phase transition temperature increased by 10.2°C for HMX-N@GNP-4h composite and 19.6°C for HMX-N@GAP-4h composite.The impact and friction sensitivity of the HMX-based composites reduced evidently in contrast to pure HMX and the physical mixture.The polymeric shell protects HMX crystals from deformation and reduces the possibility of hotspots formation in the core-shell composites.Such a route that introducing nitro,nitrate ester groups into polymer shell is certificated to possess high interfacial intensity with HMX crystals, together with enhanced polymeric stability and reduced sensitivity.So far,the grafting method mentioned for the energetic composites shows great potential for the interface design and surface modification of nitramine explosives.The influence of the core-shell structure and shell content on the mechanical properties of the composites remains to be discussed and the scale-up production of the core-shell composites is ongoing for further study.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
We gratefully acknowledge the support for this work by National Natural Science Foundation of China (Grant Nos.22175139 and 22105156).
Appendix A.Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.dt.2023.02.001.

- Defence Technology的其它文章
- Reply to the note by Li Piani et al.
- Book review:“Impact Engineering:Fundamentals,Experiments and Nonlinear Finite Elements”By Marcilio Alves(2020)Price at Amazon: US$ 85.67.www.impactbook.org
- Note on: “Ballistic model for the prediction of penetration depth and residual velocity in adobe: A new interpretation of the ballistic resistance of earthen masonry”
- Remote sensing of air pollution incorporating integrated-path differential-absorption and coherent-Doppler lidar
- Micro defects formation and dynamic response analysis of steel plate of quasi-cracking area subjected to explosive load
- Experimental and numerical study on protective effect of RC blast wall against air shock wave