
基于HPLC指纹图谱结合一测多评法的葛花质量控制研究
2024-02-26 10:05张立军张新玥张转平朱远锋吴春燕
天然产物研究与开发 2024年2期
张立军,张新玥,李 鹏,张转平*,朱远锋,刘 丽,王 磊,吴春燕
1安康市食品药品检验检测中心;2安康市天源植物提取有限公司,安康 725000;3商洛市药品检验所,商洛 726000;4北京中医药大学药学院,北京 100029
葛花(Puerariae Flos)为豆科葛属植物野葛(Puerarialobata(Willd.) Ohwi)或粉葛(PuerariathomsoniiBenth.)的干燥花蕾,又名野葛花、甘葛花、粉葛花或鹿藿花,收载于《名医别录》《滇南本草》《本草纲目》中,具有解酒醒酒、清湿热、除烦止呕的功效[1-3]。现代研究表明,葛花具有解酒毒、降血糖、降血脂、抗氧化、心肌保护及雌激素样等作用,被广泛用于解酒保肝、降血糖血脂、抗骨质疏松及调节免疫等保健产品开发的原料药[4]。化学成分研究显示,葛花中主要含有黄酮类、萜类、甾体类及香豆素类等,其中葛根素(puerarin,Pu)、大豆苷(daidzin,Da)、大豆苷元(daidzein,Dae)、染料木苷(genistin,Ge)、染料木素(genistein,Gee)、葛花苷(kakkalide,Ka)、黄豆苷(glycitin,Gl)、鸢尾苷(tectoridin,Td)、鸢尾苷元(tectorigenin,Tg)、鸢尾黄素-7-O-木糖葡萄糖苷(tectorigenin-7-O-xylosylglucoside,Tx)等为主要化学成分[4]。葛花主要分布于东南亚地区,其中在我国分布最广,陕西、四川、广西及云南等地是葛花主产区[5,6]。部颁标准和各省地方中药材标准已明确规定野葛和粉葛的花蕾作为我国葛花2个药用正品来源,为了研究区分,将前者称为葛花,后者称为粉葛花。我们课题组研究发现,二者化学成分存在明显差异。目前葛花仅有部颁标准和地方标准,品种鉴别存在混乱,质控指标仅性状、鉴别与检查,过于简单,有关完善的质量控制标准尚未见报道[7,8]。
指纹图谱是一种综合、宏观、可量化的色谱鉴定手段,该技术被广泛用于中药的质量评价,可较全面地反映中药中化学成分差异特征,从而达到甄别真伪优劣的目的[9-11]。故本研究采用HPLC建立不同产地葛花药材的指纹图谱;同时,在此基础上建立了一测多评分析方法(QAMS),用于葛花中Pu、Da、Dae、Ge、Gee、Ka、Gl、Td、Tg及Tx的含量测定,系统评价不同产区葛花药材的质量差异,为全面、系统地控制葛花质量提供实验依据,为葛花优质种质资源的选育和育种提供参考。
1 试验材料
1.1 仪器与试剂
BS224S电子分析天平(德国Sartorius公司);AS10200BT超声波清洗器(天津奥特赛恩斯仪器有限公司);Shimadzu LC-20AT、Shimadzu LC-2030高效液相色谱仪(日本岛津公司);Agilent 1200 Series型高效液相色谱仪,Agilent 5 TC C18(4.6 mm×250 mm,5 μm)色谱柱2(美国Agilent科技有限公司);Phenomenex ODS C18(4.6 mm×250 mm,5 μm)色谱柱1;Waters sunfire C18(4.6 mm×250 mm,5 μm)色谱柱3。葛根素(批号110752-201816,纯度95.40%)、大豆苷(批号111738-201904,纯度93.40%)、大豆苷元(批号111502-202003,纯度99.30%)、染料木苷(批号111709-201702,纯度99.90%)、染料木素(批号111704-202104,纯度98.80%)(中国食品药品检定研究院)。葛花苷(批号CHB201122,纯度98.00%)、黄豆苷(批号CHB201230,纯度98.00%)、鸢尾苷(批号CHB201104,纯度98.50%)、鸢尾苷元(批号CHB201219,纯度98.50%)、鸢尾黄素-7-O-木糖葡萄糖苷(批号CHB201201,纯度98.00%)均购于中国成都克洛玛生物科技有限公司。
1.2 葛花药材
20批葛花(GH)与10批粉葛花(FGH)药材分别采集于陕西、四川、广西及云南等地区(见表1),经安康市食品药品检验检测中心张转平主任药师鉴定分别为豆科葛属植物野葛(Puerarialobata(Willd.) Ohwi)和粉葛(PuerariathomsoniiBenth.)的干燥花蕾。

表1 20批葛花和10批粉葛花药材产地信息
2 方法与结果
2.1 供试品溶液制备
葛花、粉葛花分别粉碎过40目筛,取约0.2 g,精密称定,置于具塞锥形瓶中,加甲醇25 mL,称定重量,超声处理(功率300 W;频率55 kHz;水温30 ℃)30 min,放冷,用甲醇补足减失的重量,摇匀,滤过,进样前用0.45 μm微孔膜过滤,作为供试品溶液。
2.2 混合对照品溶液制备
取葛根素、大豆苷、大豆苷元、染料木苷、染料木素、葛花苷、黄豆苷、鸢尾苷、鸢尾苷元及鸢尾黄素-7-O-木糖葡萄糖苷对照品适量,精密称定,加甲醇制备质量浓度分别为8.933 3、10.266 7、11.800 0、56.333 3、11.200 0、796.666 7、74.666 7、48.000 0、14.400 0、700.000 0 μg/mL的混合对照品溶液,4 ℃冰箱中存贮备用。
2.3 色谱条件
色谱柱:Phenomenex ODS C18(4.6 mm×250 mm,5 μm);流动相A:0.2%磷酸水溶液,流动相B:乙腈;梯度洗脱:0~5 min,15%→20%B;5~17 min,20→25%B;17~25 min,25%→33%B;25~45 min,33%B;45~55 min,33%→75%B,流速1.0 mL/min;进样量10 μL,检测波长265 nm;柱温35 ℃。
2.4 葛花药材HPLC指纹图谱建立
2.4.1 方法学考察
2.4.1.1 空白溶剂考察
取甲醇按“2.3”项下色谱条件进样分析,结果表明溶剂无干扰。
2.4.1.2 精密度试验
取GH-1供试品溶液按“2.3”项下色谱条件进样分析,连续进样6次,记录色谱图。以23号峰(Ka)为参照峰,计算各色谱峰相对保留时间和相对峰面积,结果各共有峰相对保留时间和相对峰面积的相对标准偏差(relative standard deviation,RSD)均分别小于0.56%、1.2%(n=6),说明该仪器的精密度良好。
2.4.1.3 稳定性试验
取GH-1供试品溶液按“2.3”项下色谱条件进样,分别于0、3、6、9、12、24、48 h测定,以Ka参照峰,计算各共有峰相对保留时间和相对峰面积,结果各共有峰相对保留时间及相对峰面积的RSD均分别小于0.93%、1.8%,表明48 h内样品的稳定性良好。
2.4.1.4 重复性试验
取GH-1样品按“2.1”项下方法平行制备6份供试品溶液,按“2.3”项下色谱条件进样分析,以Ka参照峰,计算各共有峰相对保留时间和相对峰面积,结果各共有峰相对保留时间及相对峰面积的RSD均分别小于0.57%、1.8%(n=6),表明本法有良好的重复性。
2.4.1.5 耐用性考察
取GH-1供试品溶液,分别采用“1.1”项下3台不同HPLC仪,在3根不同品牌色谱柱条件下,按“2.3”项下色谱条件进行分析并记录其色谱图。以Ka为参照峰,各色谱峰出峰顺序均未改变,各色谱峰相对保留时间RSD均分别小于2.9%,表明本法有良好的耐用性。
2.4.2 指纹图谱建立及相似度评价
分别取葛花、粉葛花样品供试液,按“2.3”项下色谱条件进样分析,分别记录色谱图,并将数据导入“中药色谱指纹图谱相似度评价系统”(2012版),采用中位数法进行多点校正生成指纹图谱共有模式及对照指纹图谱,确定葛花、粉葛花的各自共有峰分别为18、27个(见图1)。通过与对照品比对,指认出10个共有峰(见图2)。对照指纹图谱与各批次样品图谱进行相似度评价(见表2),20批葛花和10粉葛花样品相似度均大于0.85,说明不同产地葛花和粉葛花共有峰相似度较高,药材质量较均一。
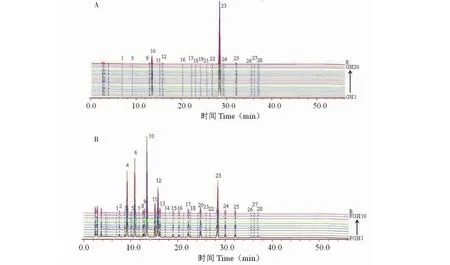
图1 葛花(A)、粉葛花(B)指纹图谱和对照图谱
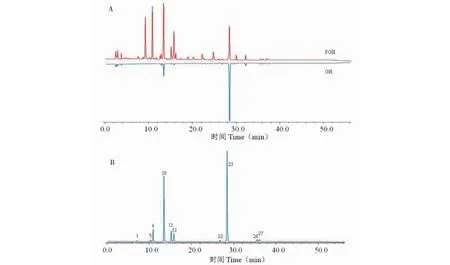
图2 葛花、粉葛花样品镜像HPLC图(A)及混合对照品的HPLC图(B)
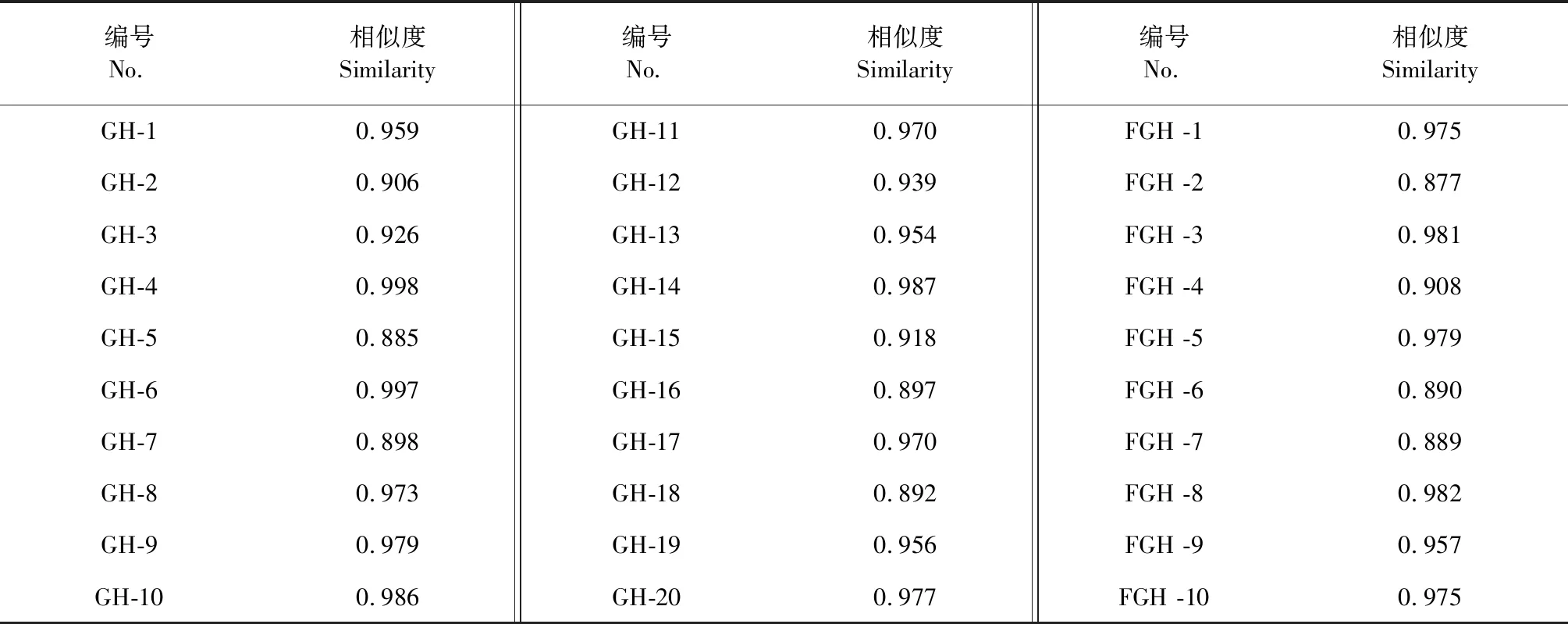
表2 20批葛花和10批粉葛花样品的相似度结果
2.5 多指标成分含量测定
为了更好地评价不同产地葛花质量,在特征图谱的基础上,建立了QAMS方法测定Pu、Da、Gl、Tx、Ge、Td、Dae、Ka、Gee、Tg 10个主要特征成分含量。其供试品制备、对照品溶液制备、色谱条件分别见“2.1”“2.2”及“2.3”项下所示。
2.5.1 精密度、稳定性及重复性试验
精密度、稳定性及重复性测定方法同“2.4.1”项,记录Pu、Da、Gl、Tx、Ge、Td、Dae、Ka、Gee及Tg 10个主要成分的峰面积,并计算其质量分数的RSD值。结果表明,各成分精密度的RSD均小于1.6%、稳定性的RSD值均小于1.7%、重复性的RSD值均小于2.0%,说明该方法稳定、可靠。
2.5.2 线性方程、定量限与检测限
精密吸取混合对照品贮备液0.25、0.5、1.0、1.5、2.5 mL,用甲醇定容至5.0 mL,摇匀。分别按“2.3”项下色谱条件进样分析,测定峰面积,以峰面积(Y)对质量浓度(X)进行线性回归。将对照品溶液进行逐级稀释,分别以信噪比的3倍和10倍计算检测限与定量限,见表3。
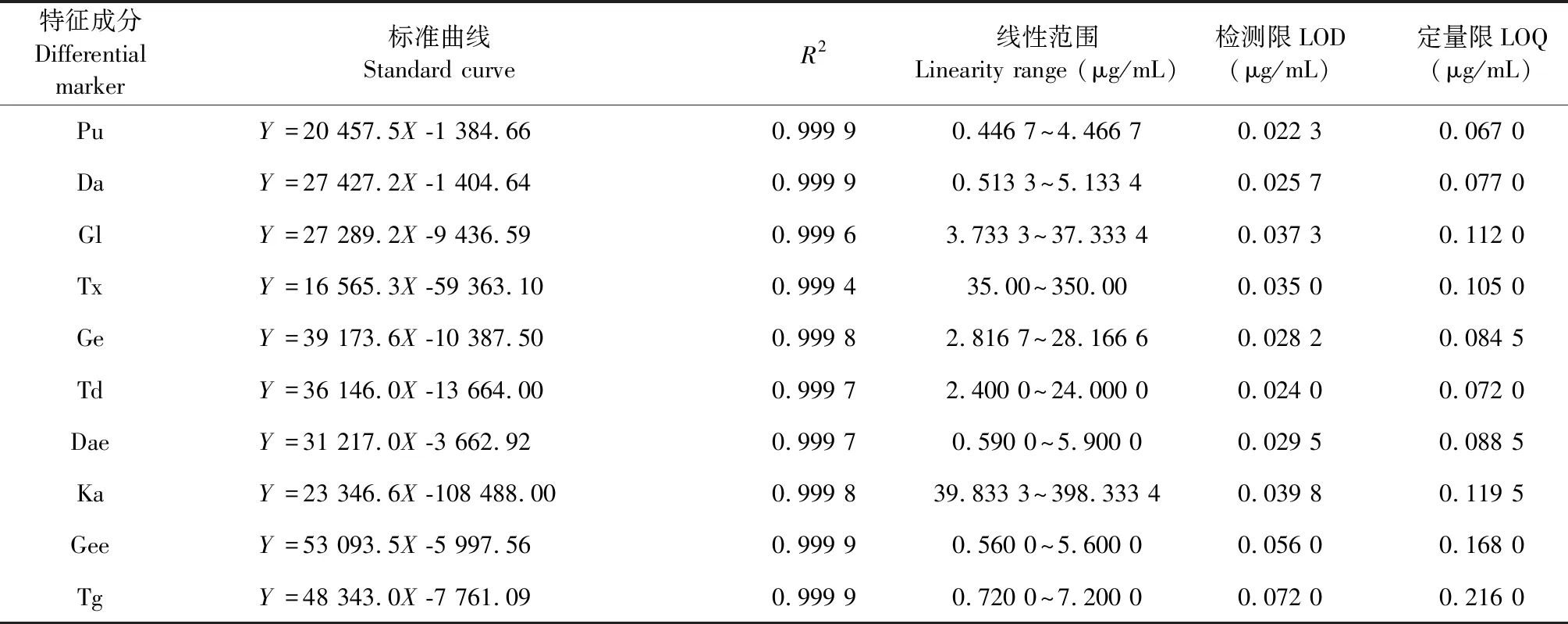
表3 10种葛花黄酮类成分的线性关系、定量限与检测限考察
2.5.3 加样回收率试验
取已知含量的GH-1样品粉末0.1 g共6份,精密称定,分别置于精密加有“2.2”项下2.00 mL混合对照品的干燥具塞锥形瓶中,按“2.1”项下方法制备供试品溶液,按“2.3”项下色谱条件进样分析,计算Pu、Da、Gl、Tx、Ge、Td、Dae、Ka、Gee及Tg的平均回收率,回收率范围在95.22%~105.7%,RSD均小于2.9%,表明方法的回收率良好。
2.5.4 一测多评方法(QAMS)建立
2.5.4.1 相对校正因子(f)计算
精密吸取“2.6.2”项下各混合对照品溶液适量,按“2.3”项下色谱条件进样分析,以Ka为参照物,按如下公式计算Pu、Da、Gl、Tx、Ge、Td、Dae、Gee及Tg的相对校正因子(f)[11,12],结果见表4。
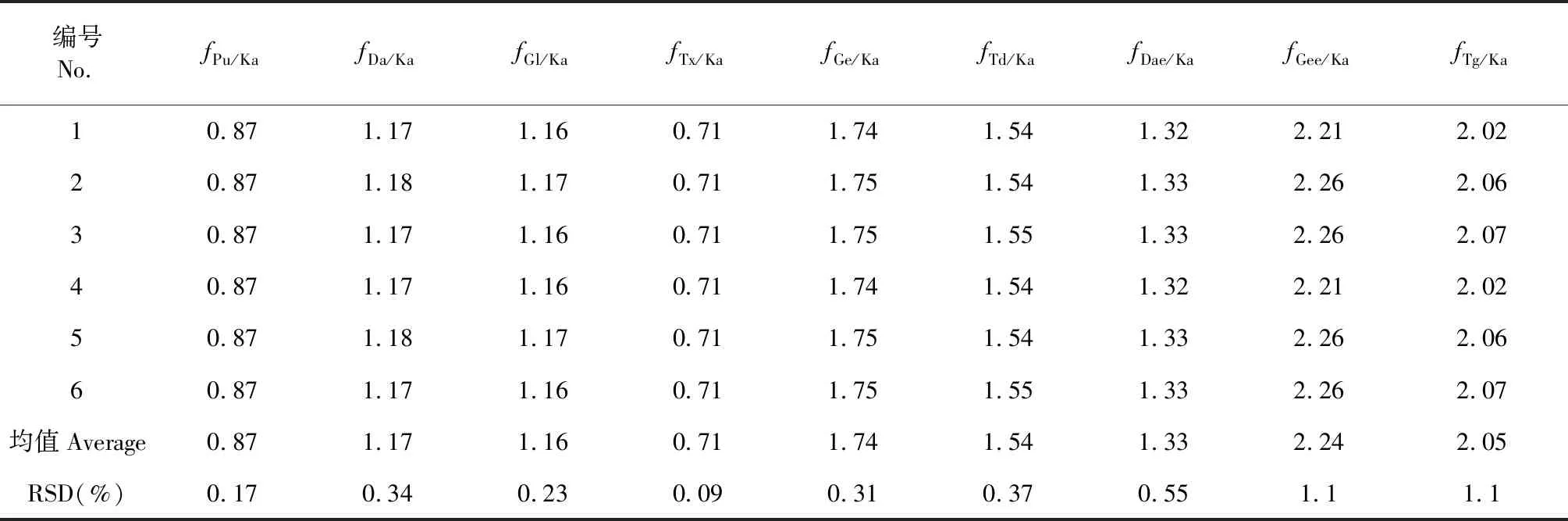
表4 相对校正因子(f)测定结果
f=fi/fs=(Ai/Wi)/(As/Ws)
(1)
式中,Ai和As分别表示待测化合物和参照化合物的峰面积(或峰高);Wi和Ws分别为待测化合物与参照化合物的质量浓度。
2.5.4.2 耐用性考察
不同高效液相色谱仪和色谱柱对f值的影响:采用“1.1”项下3种不同HPLC仪,分别考察了3种不同品牌的色谱柱对f值的影响,结果显示3种品牌色谱柱在不同HPLC仪上所测定的f的RSD均小于3.0%,表明葛花中10个黄酮成分在不同色谱系统及不同色谱柱上的耐用性较好(见表5)。
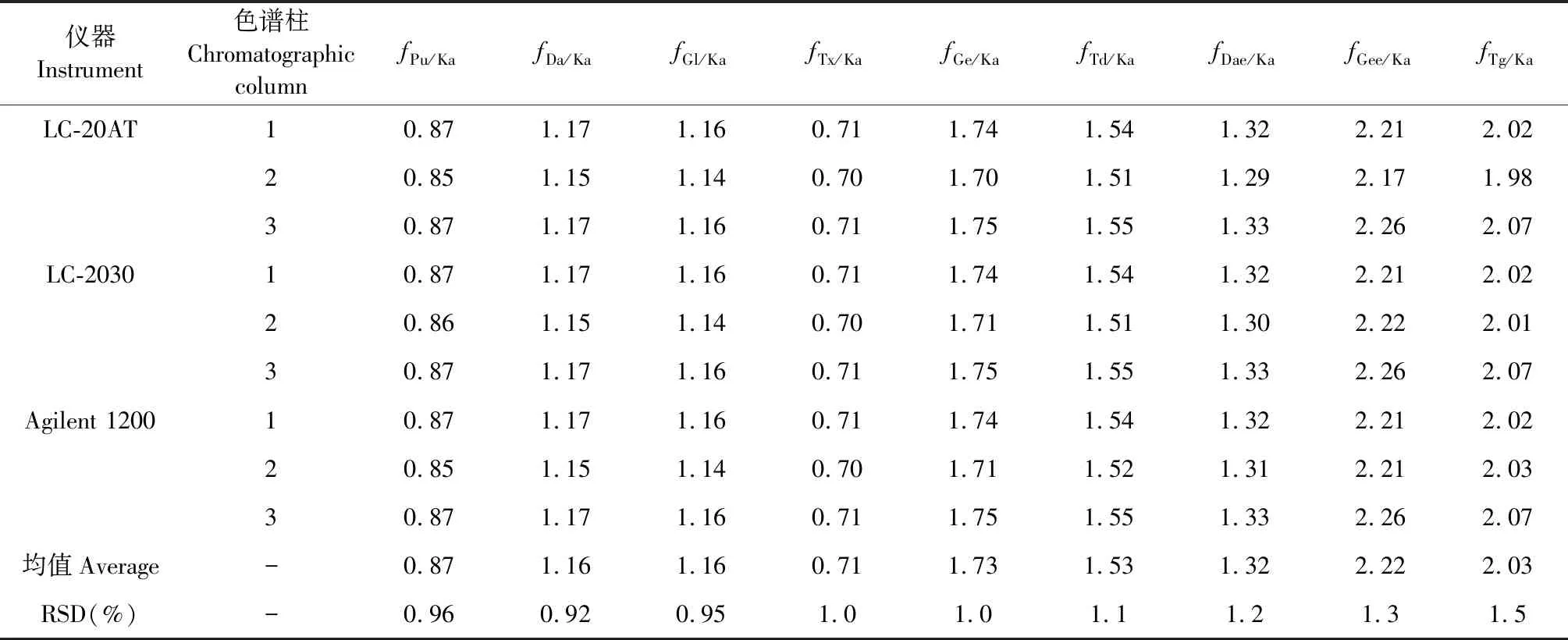
表5 不同色谱柱和HPLC系统对f的影响
不同色谱柱温对f值的影响:实验采用LC-20AT HPLC仪,色谱1号柱,考察不同柱温(30、35、40 ℃)对f的影响,结果显示各待测成分f的RSD 均小于3.0%,表明葛花中10个黄酮成分在柱温为30~40 ℃时耐受性较好(见表6)。
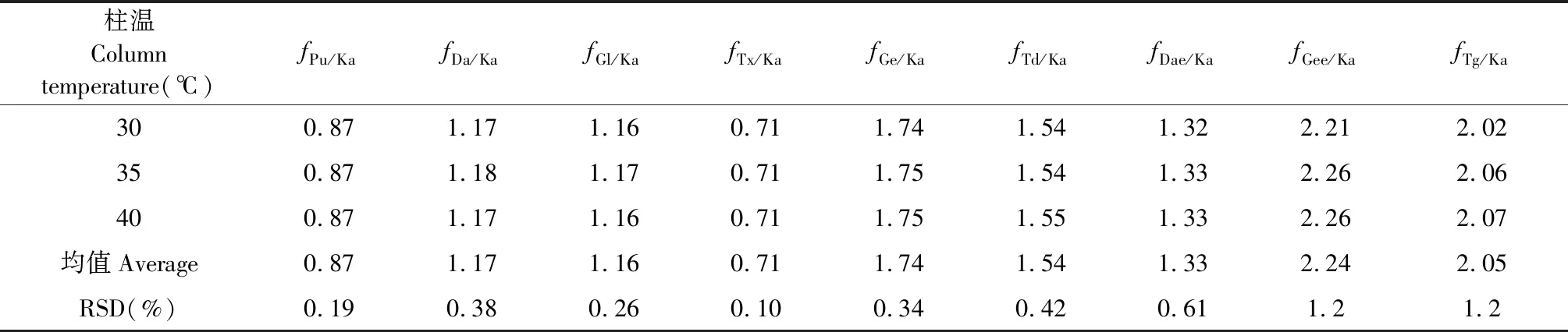
表6 不同柱温对f的影响
不同流速对f值的影响:实验采用LC-20ATHPLC仪,色谱1号柱,考察了不同流速(0.8、1.0、1.2 mL/min)对f的影响,结果显示各待测成分f的RSD 均小于3.0%,表明葛花中10个黄酮成分在流速0.8~1.2 mL/min时耐受性较好(见表7)。
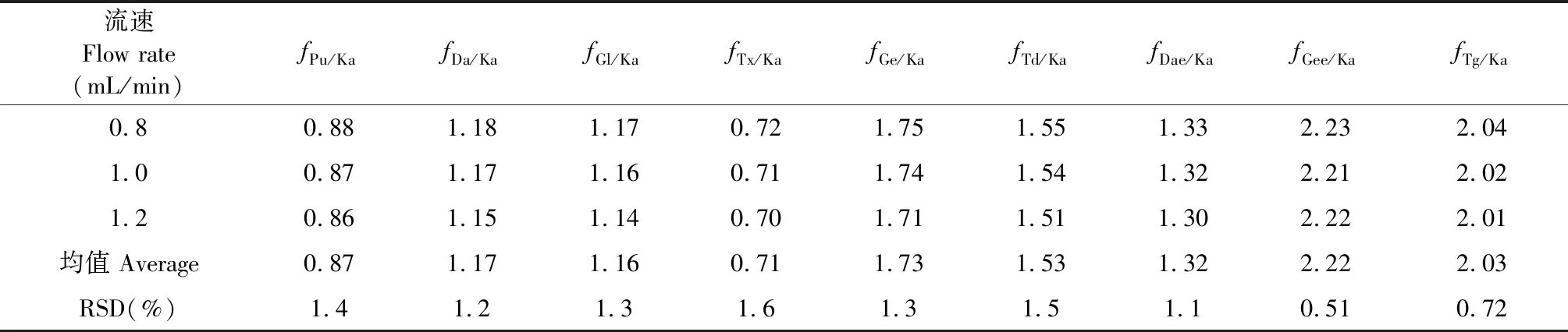
表7 不同流速对f的影响
2.5.4.3 待测成分色谱峰的定位
在QAMS的运用中,目前常用的色谱峰定位方法有相对保留时间(Rt)法、保留时间差(Δt)法、时间校正法、对照提取物法等[12]。Rt计算公式:Rti/s=ti/ts。Δt计算公式:Δti/s=ti-ts。采用“1.1”项下HPLC仪,分别考察了3根不同品牌色谱柱条件下各待测成分的Rt及Δt。结果发现,不同仪器和不同色谱柱对Δt法的影响较大,RSD>7.0%;相对而言,Rt法的差异较小。本实验采用Rt法对待测组分色谱峰进行定位,结果Pu、Da、Gl、Tx、Ge、Td、Dae、Gee及Tg相对于Ka的Rt均值分别为0.245、0.359、0.382、0.473、0.533、0.553、0.940、1.251及1.271,RSD≤2.586%,表明采用待测组分与参照峰Rt值来定位具有可行性(见表8)。
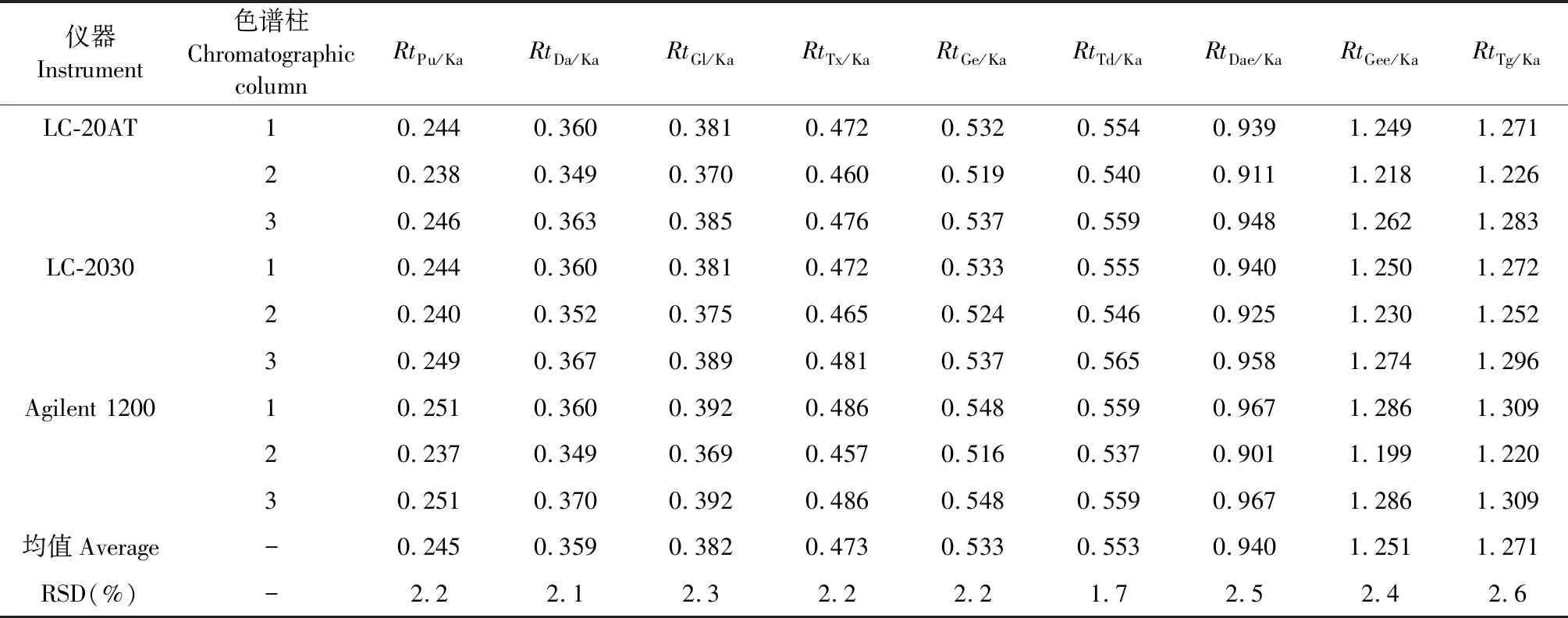
表8 不同仪器、不同色谱柱对Rt的影响
2.5.4.4 QAMS和外标法测定结果比较
分别随机从陕西抽取葛花8批、粉葛花2批;四川抽取葛花1批;云南抽取粉葛花1批;广西抽取葛花1批、粉葛花2批,按“2.1”项下供试品制备方法,“2.3”项下色谱条件进样分析,分别采用QAMS与ESM计算各成分含量,QAMS定量计算公式如下[11,12]。
(2)
式中,Ai和As分别表示待测化合物和参照化合物的峰面积(或峰高);Wi和Ws分别为待测化合物与参照化合物的质量浓度;f为待测标志物i的相对校正因子。
将QAMS计算值与外标法实测值进行配对t检验,结果P值均>0.05,表明两种方法测定的含量结果无显著性差异;进一步以相对误差(relative deviation,RD)对两种方法测定结果进行准确性评价[11]。
(3)
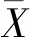
计算结果,除了FGH中Dae、Gee及Tg含量的RD小于9.0%外,其余的RD均小于5.0%,表明2种测定方法的结果无显著性差异(见表9)。
同时对外标法测定结果进行聚类热图分析,结合SPSS 24.0软件对其进行独立样本t检验分析,结果显示葛花、粉葛花在成分种类和含量存在明显的差异,其中葛花中Ka、Dae、Gee含量显著高于粉葛花(P<0.01),未检测到Da、Gl;粉葛花中Tx、Td、Ge含量显著高于葛花(P<0.01);聚类热图可将葛花、粉葛花分为2大类。不同产地来源的葛花、粉葛花药材中10个成分含量均存在较大差异,其中位于秦巴山区安康旬阳、紫阳、汉中镇巴及商洛镇安等县的葛花各成分含量较高,质量较优;位于广西、云南等地粉葛花各成分含量较高,质量较优。研究结果清晰地表明了不同产地葛花药材的质量差异(见表9和图3)。

图3 葛花、粉葛花差异性标志物含量聚类热图
3 讨论与结论
实验采用二极管阵列检测器,对10个黄酮类成分进行了200~400 nm全波长扫描,结果显示Pu、Da、Dae、Ge、Gee、Ka、Gl、Td、Tg及Tx 10个成分的紫外光谱图较为相似,均在256~274 nm波长处有最大吸收。选择在265 nm波长条件下,分析发现10种待测成分具有良好的基线分离,且基线平稳,可以满足定量分析的要求。实验考察了75%乙醇;95%乙醇;75%甲醇及甲醇各25 mL作为提取溶剂,结果选择甲醇作为提取溶剂时,色谱峰数目适宜,便于分离。实验也考察了超声及热回流处理方式,最终选择超声30 min,方法简便且效率高。实验还考察了甲醇-水、乙腈-水、乙腈-0.2%磷酸水溶液及乙腈-0.2%甲酸水溶液等流动相体系。最终选择乙腈-0.2磷酸水溶液梯度洗脱体系,分离效果好,拖尾得到很好的改善。
为有助于葛花全面质量控制,实验分别选择了活性明确、含量较高的各类代表性成分作为QAMS法含量测定的指标成分,主要包括Pu、Da、Dae、Ge、Gee、Ka、Gl、Td、Tg及Tx等为主要化学成分[4,13]。从各成分含量测定结果、各成分色谱分离情况、对照品价格与易得情况看,Ka含量较高,分离效果好,在谱图中容易识别,处于色谱图的中位,利于其他成分色谱峰的相对保留时间计算和色谱峰的识别,且Ka性质稳定、价廉易得,具有降血脂血糖、改善微循环及激活乙醇脱氢酶的活性[14,15]。因此,本研究选择Ka作为QAMS法含量测定的参照物。
本实验建立的指纹图谱可很好表征葛花中化学成分的信息,在指纹图谱基础上建立的QAMS法可有效测定葛花、粉葛花中主要有效成分Pu、Da、Gl、Tx、Ge、Td、Dae、Ka、Gee及Tg的含量,可全面反映葛花药材的整体质量信息。根据含量测定结果与聚类热图可直观看出,葛花、粉葛花分为2大类,来自不同产区的20批葛花、10批粉葛花药材中主要有效成分种类及含量差异很大。差异成分为Ka、Da、Gl、Tx、Td及Ge。其原因与葛花和粉葛花来源不同有关。
葛花目前收录野葛和粉葛的干燥花蕾共同作为药材来源,鉴于葛花和粉葛花中主要有效成分存在较大差异,两者的临床疗效应该也有所不同,因此,在产品开发和临床应用中应当加以区分使用。同时葛花具有较大保健产品开发前景,亟需建立规范的葛花质量评价体系。本研究所建立的指纹图谱结合一测多评方法在减少对照品使用的情况下,可实现葛花药材的多成分、多指标质量控制;同时能很好地区分葛花、粉葛花药材,并且能对不同产地葛花药材进行有效的质量评价;实验结果可为葛花质量标准修改完善提供参考依据,可以为葛花优质种质资源的选育和育种提供参考,在中药整体质量评价模式中具有良好的应用前景。
猜你喜欢
保健与生活(2023年17期)2023-10-16
保健与生活(2023年19期)2023-08-15
小哥白尼(趣味科学)(2021年11期)2021-02-28
小天使·一年级语数英综合(2020年10期)2020-12-16
源流(2020年6期)2020-08-03
农家之友(2020年2期)2020-05-19
中成药(2018年8期)2018-08-29
中成药(2017年6期)2017-06-13
浙江农业学报(2016年7期)2016-06-15
自动化学报(2016年8期)2016-04-16
