
新生儿型氨甲酰磷酸合成酶Ⅰ缺乏症合并地中海贫血1例及文献复习
2024-02-20 05:42柯海燕邹甜甜费世暖
检验医学与临床 2024年3期
柯海燕,邹甜甜,雷 琳,费世暖,江 鸿,李 胜
1.湖北理工学院附属妇幼保健院/黄石市妇幼保健院医学检验科,湖北黄石435000;2.出生缺陷防治黄石市重点实验室,湖北黄石435000;3.湖北理工学院附属妇幼保健院/黄石市妇幼保健院儿童保健科,湖北黄石435000;4湖北理工学院附属妇幼保健院/黄石市妇幼保健院儿科,湖北黄石435000
新生儿型氨甲酰磷酸合成酶Ⅰ(CPSⅠ)缺乏症(CPSⅠD)是尿素循环障碍疾病中比较罕见的一种,属于常染色体隐性遗传病,呈高度遗传异质性,与CPSⅠ基因有关,基因变异位于2号染色体,在新生儿期发病极为凶险[1-3]。世界范围内CPSⅠD发病率为1/10万~1/80万,有种族和地区差异,症状特异性不强,临床极易误诊,但我国发病率尚不明确[4]。我国新生儿筛查中心7 819 662 名新生儿筛查结果显示,CPSⅠ 缺乏症发病率为 1/1 954 916,其他临床研究均为个例报道[5-7]。地中海贫血属常染色体隐性遗传,是由于珠蛋白链合成障碍导致的溶血性贫血,也是人类最常见的单基因遗传病之一[8-10]。地中海贫血属于遗传性溶血性疾病,该部分患者因为血红蛋白肽链基因缺失或者突变,导致体内血红蛋白水平低于正常水平,最终出现贫血症状[11]。本文分析了1例新生儿期CPSⅠD合并地中海贫血的临床特征及基因变异情况以供临床参考。
1 临床资料
1.1一般资料 患儿,男,出生7 d,因吮奶差、拒奶、反应低下于2019年7月4日下午转至湖北理工学院附属妇幼保健院。出生体质量3.2 kg,否认窒息史,混合喂养,出生2 d开始出现吮奶差,随后出现精神反应差,拒奶,无发热及抽搐,无呕吐及呛奶,2019年7月2日于外院住院,经抗感染、光疗、肝酶诱导剂、护脑、部分静脉营养对症支持治疗后仍反应低下,医生会诊后以“新生儿反应低下”转入本院。
1.2入院体格检查 体温36.2 ℃,呼吸46次/分,脉搏131次/分,血压72/40 mm Hg,体质量2.88 kg,身长50 cm,精神反应差,营养发育正常,前卤平软,皮肤黄染(+++),唇周微绀,呼吸欠规则,无吐沫,双肺呼吸音粗,无啰音,心音有力,腹软,下腹部可触及4 cm×4 cm包块,质稍硬,肠鸣音正常,脐部无渗血,四肢肌张力增高,未引起病理反射。
1.3实验室检查 2019年7月4日实验室检查结果显示,白细胞计数22.52×109/L↑,中性粒细胞百分比52.6%,淋巴细胞百分比35.8%,单核细胞百分比10.4%↑,红细胞计数6.99×1012/L↑,红细胞比容57.9%↑,血红蛋白水平193 g/L↑,血小板计数438×109/L↑,C反应蛋白及网织红细胞计数均正常;血氨543 μg/dL↑。隔天查血浆氨>1 000 μg/dL↑;血气分析pH值7.58↑,二氧化碳分压14 mm Hg↓,氧分压87 mm Hg,实际碳酸氢根13.1 mmol/L↓,标准碳酸氢根21.6 mmol/L↓,剩余碱-4.4 mmol/L↓,红细胞外液剩余碱-8.8 mmol/L↓,二氧化碳总量13.5 mmol/L↓,氧饱和度92 mmol/L,总血红蛋白17.5 g/dL,红细胞比容 53%↑,钾3 mmol/L↓,钠129 mmol/L↓,离子钙0.9 2mmol/L↓,乳酸6.5 mmol/L↑,葡萄糖4.2 mmol/L。地中海贫血血红蛋白毛细管电泳结果和外显子测序法验证地中海贫血基因型为SEA杂合。
1.4影像学检查 磁共振成像检查未见明显异常。
1.5其他检查 由于患儿血氨短期内急剧升高,高度怀疑为遗传代谢疾病,进一步行血串联质谱分析,瓜氨酸为1.5 μmol/L↓。患儿外显子测序结果显示为2个CPSⅠ基因变异,见表1。串联质谱检测结果均是鸟氨酸轻度降低,3-羟基异戊酰基肉碱轻度升高,其余正常;血氨均轻度升高。
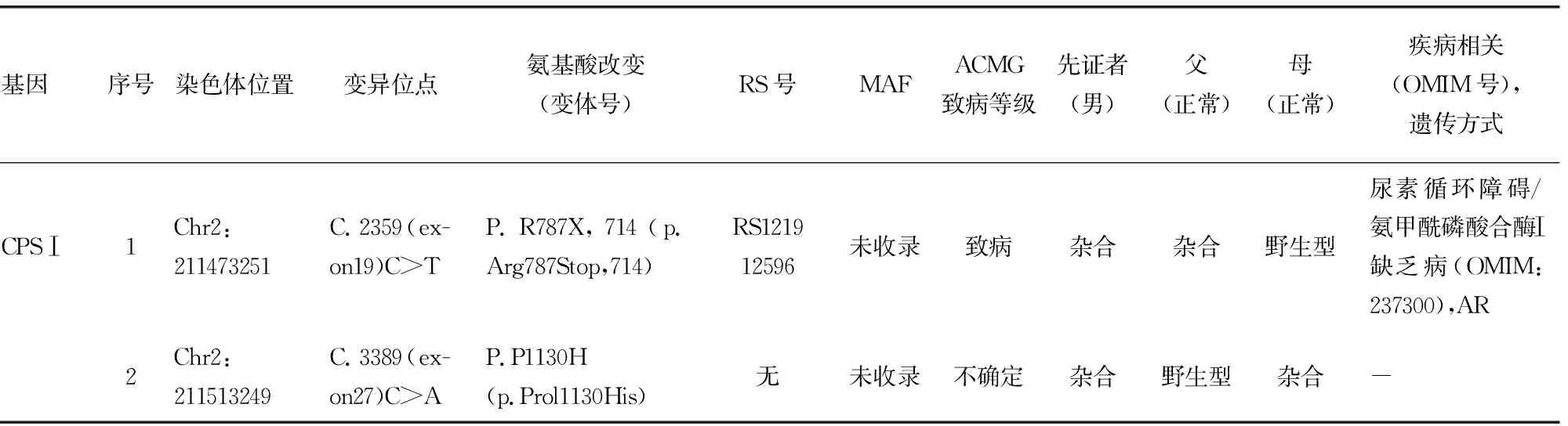
表1 先证者(患儿)及其家属的致病基因位点列表
2 讨 论
CPSⅠ催化CO2和NH3合成氨甲酰磷酸,是尿素循环的第一个步骤,酶缺乏使氨在体内水平增加产生高氨血症,临床表现多变,发病年纪与酶缺陷程度有关,新生儿期发病多见,病死率极高[12-14]。CPSⅠD根据发病年龄、酶活性降低和临床表现分为2个独立表型:新生儿型和迟发性,由于该病发病罕见,症状呈现非特异性,临床容易误诊[15]。本研究患儿出生后2 d出现吮吸差,随后进展为精神反应差,拒奶,嗜睡,昏迷,早期出现黄疸并合并肺炎,发病早期白细胞升高易误诊为炎症,多次血气检查提示呼吸性碱中毒,可能是血氨升高刺激中枢系统导致过度通气所致,血氨由543 μg/dL急剧升至大于1 000 μg/dL,串联质谱提示瓜氨酸水平显著降低,考虑为尿素循环障碍。
进一步检查全外显子基因测序提示CPSⅠ基因变异的致病性和不确定变异位点,分别来自父亲的C.2359(exon19)C>T的无义变异(序号1)和母亲的C.3389(exon27)C>A错义变异(序号2)。根据《遗传变异分类标准与指南》[16]相关原则,序号1变异有PVS1、PS1、PM2、PP3等阳性特性,生物学致病等级判断为致病;序号2变异有PM2、PM3、PP3等阳性特性,生物学致病等级判断为不确定。序号1的MAF(dbSNP数据库)为0.000 016,即该变异的杂合形式在人群的分布率为0.001 6%,说明该变异属于罕见的致病变异;序号2变异MAF(dbSNP数据库)未收录,且这2个变异在千人基因组、EXAC数据库等均未收录。
CPSⅠD患儿在新生儿期出现喂养困难、嗜睡、呕吐、惊厥、昏迷等症状易误判为感染、缺血缺氧性脑病,但患儿多在尚未确诊前已死亡,是疾病诊断的难点。故临床需与以下疾病鉴别诊断:(1)大量氨基酸输液者;(2)婴儿期暂时性的高氨血症;(3)乙酰谷氨酸合成酶缺乏者;(4)鸟氨酸甲酰转移酶缺乏症;(5)Rey综合征;(6)有机酸血症,必要时需肝活检特异性、酶活性以确诊。CPSⅠD引起的高氨血症严重危及患者的生命安全和认知能力,原则上可通过降低血氨水平进行治疗,包括限制蛋白质摄入,补充一定瓜氨酸、精氨酸,血氨显著升高时可进行血液或腹膜透析,肝脏移植是治疗的重要手段,且移植术后能获得长期生存[17]。
该患儿出生后多次因病住院,临床特征:易怒,智力障碍,癫痫发作,共济失调,昏睡,昏迷,全面性发育迟缓,卒中,生长不良,呼吸性碱中毒,发作性氨中毒,高氨血症,呕吐。治疗以支持治疗为主,移除氨及代谢产物,消除分解代谢,后期需透析治疗,伴有感染、呼吸衰竭等情况,予以抗感染、呼吸支持及对症处理,预后极差,存活率低,提示尿素循环障碍/CPSⅠD,系尿素循环障碍及高氨血症,属基因代谢性疾病。
2020年2月21日患儿再次入院时仍在抽搐,伴有呻吟,血氧饱和度50%,血压测不出,立即气管插管机械通气,予以镇静止惊、脱水降低颅内压、纠酸、强心、扩容、改善循环、抗炎及对症支持治疗,并辅以灌肠通便。告知家属病情后,家属要求放弃呼吸支持、输液及相关抢救措施,最终患儿宣告临床死亡。
本例CPSⅠD患儿同时合并轻型地中海贫血,起病急进展快,临床表现复杂,出生不久便出现持续性轻中度贫血,但血小板持续升高原因未明,值得进一步探讨研究。总之,常规的新生儿疾病筛查技术结合精准的基因测序可为疾病的早发现早诊断提供重要支持。
猜你喜欢
首都食品与医药(2021年5期)2021-03-22
转化医学电子杂志(2018年11期)2018-01-16
中国卫生(2016年12期)2016-11-23
股市动态分析(2016年17期)2016-10-20
股市动态分析(2016年13期)2016-10-17
股市动态分析(2016年10期)2016-09-30
股市动态分析(2016年2期)2016-09-27
当代经济(2016年26期)2016-06-15
中国现代药物应用(2016年10期)2016-03-06
江苏年鉴(2014年0期)2014-03-11
