
STAT3 ameliorates truncated tau-induced cognitive deficits
2024-02-11 08:39BinggeZhangHualiWanMaimaitijiangMaierwufuQianLiuTingLiYeHeXinWangGongpingLiuXiaoyueHongQiongFeng
中国神经再生研究(英文版) 2024年4期
Bingge Zhang,Huali Wan,Maimaitijiang MaierwufuQian LiuTing LiYe HeXin WangGongping LiuXiaoyue Hong,Qiong Feng
Abstract Proteolytic cleavage of tau by asparagine endopeptidase (AEP) creates tau-N368 fragments,which may drive the pathophysiology associated with synaptic dysfunction and memory deterioration in the brain of Alzheimer’s disease patients.Nonetheless,the molecular mechanisms of truncated tau-induced cognitive deficits remain unclear.Evidence suggests that signal transduction and activator of transcription-3 (STAT3) is associated with modulating synaptic plasticity,cell apoptosis,and cognitive function.Using luciferase reporter assays,electrophoretic mobility shift assays,western blotting,and immunofluorescence,we found that human tau-N368 accumulation inhibited STAT3 activity by suppressing STAT3 translocation into the nucleus.Overexpression of STAT3 improved tau-N368-induced synaptic deficits and reduced neuronal loss,thereby improving the cognitive deficits in tau-N368 mice.Moreover,in tau-N368 mice,activation of STAT3 increased N-methyl-D-aspartic acid receptor levels,decreased Bcl-2 levels,reversed synaptic damage and neuronal loss,and thereby alleviated cognitive deficits caused by tau-N368.Taken together,STAT3 plays a critical role in truncated tau-related neuropathological changes.This indicates a new mechanism behind the effect of tau-N368 on synapses and memory deficits.STAT3 can be used as a new molecular target to treat tau-N368-induced protein pathology.
Key Words: Alzheimer’s disease;apoptosis;cognitive deficit;memory;neurodegenerative disease;neuron loss;N-methyl-D-aspartic acid receptor;STAT3;synapse;tau-N368
Introduction
Alzheimer’s disease (AD) is a neurological condition that gradually destroys brain cells over time.Its pathological hallmarks include neurofibrillary tangles formed by hyperphosphorylated tau,and senile plaques comprising extracellular accumulation of amyloid-β peptide (Grundke-Iqbal et al.,1986;Ju and Tam,2022).Tau protein is a major microtubule-associated protein that promotes axon transport and maintains neuronal function by aiding microtubule assembly and stability (Alonso et al.,1994;Ebneth et al.,1998;Audouard et al.,2015).However,tau hyperphosphorylation under disease conditions promotes its aggregation and mislocalization and can induce neurodegeneration in related tauopathies (Alonso et al.,1994;Wegmann et al.,2018).In addition to phosphorylated tau,fragmented tau also performs a critical role in tau pathogenesis (Hrnkova et al.,2007;Al-Hilaly et al.,2020;Pollack et al.,2020;Vogels et al.,2020;Carlomagno et al.,2021).Recent research indicates that the presence of fragmented tau in cerebrospinal fluid promotes the secretion of tau (Kim et al.,2010).One possible contributor to the onset and progression of AD is truncated tau.
Asparagine endopeptidase (AEP,also named δ-secretase) is a lysosomal cysteine protease activated in AD brain.This age-dependent enzyme cleaves protein substrates on the C-terminal side of asparagine,while cleaving the amyloid precursor protein at residues N373 and N585 and directly cleaving tau at two sites,N255 and N368 (Zhang et al.,2014,2015).Tau cleavage at position N368 by AEP eliminates the protein’s ability to bind to microtubules,thus enhancing its capacity to aggregate and increase its neurotoxicity.Tau1-368 (tau-N368) fragment exhibits stronger aggregation ability and neurotoxicity than full-length tau.Overexpression of the tau1-368 fragment in C57BL/6 mouse brain induced AD-like pathological changes and behavioral disorders (Zhang et al.,2021).Moreover,inactivation of AEP inhibited the formation of tau-N368 fragments,reduced tauopathies in tau P301S,and rescued the cognitive defects in AD models (Zhang et al.,2015,2017b).Chronic treatment with specific small-molecule inhibitors of AEP attenuated tau cleavage and ameliorated synaptic dysfunction and cognitive impairments in transgenic mouse models (Zhang et al.,2016,2017b).In addition,patients with AD were shown to have an increase in tau-N368 levels in both their brain tissue and cerebrospinal fluid (Zhang et al.,2015;Blennow et al.,2020;Simrén et al.,2021).Together,these data show that truncated tau-N368 is significantly involved in the onset and progression of AD.Growing evidence shows that signal transducer and activator of transcription (STAT) plays an integral role in the pathological process of AD (Levy and Darnell,2002;Chiba et al.,2009;Tai et al.,2011;Park et al.,2013;Yu et al.,2014).Once activated,as an intranuclear transcription factor,STATs form dimers in the nucleus and participate in regulating gene expression.STATs are implicated in numerous biological processes,such as cell proliferation,differentiation,immunological modulation,cell survival,and apoptosis (Yu et al.,2009;O’Shea and Plenge,2012;Li et al.,2015;Zhang et al.,2017a;Damasceno et al.,2020).Recently,our previous work (Li et al.,2019;Hong et al.,2020;Wan et al.,2021) showed that STAT1 acted to suppress N-methyl-D-aspartic acid receptor (NMDAR) expression and induce synapse and memory deficits in overexpressing P301L-htau or htau AD mouse models.The accumulation of htau or P301L-htau inhibited STAT3 translocation into the nucleus by inducing acetylation of STAT1 and enhancing the interplay between STAT1 and STAT3 within the cytoplasm.Thus,overexpressing STAT3 or non-acetylated STAT1 ameliorated the htau or P301L-htau-induced synaptic plasticity impairments and cognitive deficits by upregulating NMDAR expression (Li et al.,2019;Hong et al.,2020;Wan et al.,2021).The STAT family,including STAT1 and STAT3,are involved in tauopathies (Lim and Cao,2006;Yamada et al.,2008;Nicolas et al.,2012;Hsu et al.,2014).Therefore,we postulated that STAT3 also performs a fundamental function in the cognitive impairments caused by tau-N368.This study aimed to investigate whether STAT3 could ameliorate the cognitive deficits caused by truncated tau accumulation and the underlying mechanisms.
Methods
Animals
There are large cyclical fluctuations in physiological indicators and unstable hormone levels in female C57BL/6 mice (ter Horst et al.,2012);therefore,female mice were excluded from the present study to aid interpretation of the data.Animals used in this study were male C57BL/6 mice,weighing 20-25 g and aged 8 weeks (n=40) obtained from the Cavens Biogle (Suzhou) Model Animal Research Co.,Ltd.,(Suzhou,China;license No.SCXK (Su) 2018-0002).Each animal had an unlimited supply of water and food and was housed at a constant temperature of 24 ± 2°C with 55 ± 5% humidity and a 12-hour light/dark cycle each day.Research involving animals was carried out following the ‘Policies on the Use of Animals and Humans in Neuroscience Research’,which were revised and approved by the Society for Neuroscience in 1995,as well as the Guidelines for the Care and Use of Laboratory Animals of the Ministry of Science and Technology of China,and the Institutional Animal Care and Use Committee at Tongji Medical College.The Academic Review Board of Tongji Medical College,Huazhong University of Science and Technology granted approval for the animal experiments on May 1,2021 (approval No.2021-3192).
The mature mice (n=40) were randomly divided into four groups.In the vector (VEC) group (n=10),mice were stereotaxically injected with adenoassociated virus (AAV)-eGFP virus.In the STAT3 group (n=10),mice were stereotaxically injected with AAV-STAT3 virus.In the tau-N368 group (n=10),mice were stereotaxically injected with AAV-eGFP-htau-N368 virus.In the tau-N368+STAT3 group (n=10),mice were stereotaxically injected with AAVSTAT3 and eGFP-htau-N368 virus.All the procedures were performed in a double-blind manner.The study procedure is shown inAdditional Figure 1.
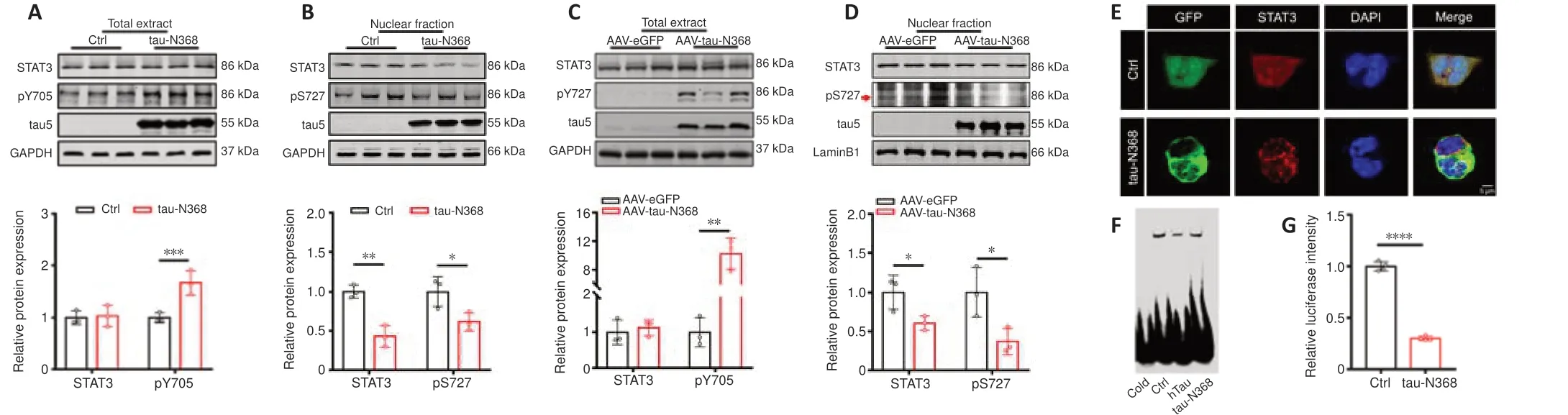
Figure 1|Overexpression of tau-N368 fragment inactivated STAT3 by inhibiting nuclear translocation.
Stereotaxic brain injection
AAV-eGFP-htau-N368 (1.5×1013vector genomes (v.g.)/mL),control AAVeGFP (1.2×1013v.g./mL),and AAV-STAT3 (1.2×1013v.g./mL) were driven by a cytomegalovirus promoter,and sourced from OBiO Biologic Technology Corp.,Ltd.(Shanghai,China).Mice at 2 months old were given a 0.10 µL/minute bilateral stereotactic injection of virus into the CA3 area of the hippocampus (anterior-posterior ±2.0 mm,medial-lateral -1.5 mm,dorsal ventral -2.0 mm;Franklin and Paxinos,2007).There was a 15-minute wait after the syringe with the needle was inserted before it was removed.Neither the animals’ regular activity nor their mortality rate changed noticeably because of treatment.
Morris water maze
The Morris water maze assessment was performed to detect spatial learning and memory abilities around 4 weeks after the brain was infused with virus (Hong et al.,2020).The instrument (Tymon Software Co.,Chengdu,China) consisted of a circular tank filled with water (23 ± 2°C) measuring 50 cm in height and with a diameter of 120 cm.The water maze was separated into four distinct quadrants;the walls of the pool in each quadrant were marked with distinctive drawings to guide the mice.Central to the third quadrant of the pool,a hidden platform measuring 10 cm in diameter was placed roughly 2 cm under the surface of the water.To evaluate spatial learning,the animals were trained to locate the hidden platform over 6 successive days (between 13:00 to 19:00),with three trials each day separated by a 30-second interval.For every training test,the mice were placed gently into the maze facing the pool’s wall from each of the three remaining quadrants (not the quadrant with the platform),and then allowed 1 minute to search for the platform.The latency (to reach the platform) was recorded for 60 seconds.If the mice took longer than 1 minute to locate the platform,they were guided to the platform for 30 seconds.Once the training was complete and the platform was withdrawn (on day 7),a test of spatial memory was conducted.A ceilingmounted video camera located 1.5 m above the water’s surface was used to record the latency (first time finding the target platform area),total count of platform crossings,percentage of time spent on the platform’s quadrant,and swimming speed.
Barnes maze test
The Barnes maze test was carried out at 4 weeks after the brain was injected with virus (Mei et al.,2021).A circular platform with a diameter of 120 cm was used to construct the maze.There were 20 holes measuring 50 mm in diameter within the 25 mm edge of the platform.A passageway leading through one of these holes opened into a dark compartment,referred to as the target box.Additionally,120 W lights were positioned along the room’s perimeter,with their upward-facing bulbs pointing towards the ceiling.Mice were guided to locate the target box for 4 days in a row as part of a spatial learning experiment.Each training session lasted for 3 minutes,and there were two sessions each day separated by an interval of 15 minutes.In between each phase of the experiment,the platform and target box were wiped with 70% ethanol.Spatial memory was assessed on day 5 following training,and the target box was withdrawn.During each trial,the amount of time that it took to find the target box and the number of mistakes made before reaching the box were recorded.
Novel object recognition test
The novel object recognition test was carried out at 4 weeks after the brain was injected with virus (Hong et al.,2020).One day before the beginning of the test,the mice were given 5 minutes to become accustomed to an empty arena (a 50 cm3plastic container).During the first day of training,two identical objects A and B were put at opposite corners of the arena,and the mice were given 5 minutes to investigate.The amount of time spent inspecting objects A and B was recorded.The next day,object B was swapped for a new object C,which was of the same material and size but different in shape.The mice were given another 5 minutes to investigate the new item.Between each habituation period,70% ethanol was used to clean the area and objects.During the test,a camera was set up above the arena to film the behavior.The preferential index was derived as the amount of time spent inspecting object C/amount of time spent inspecting objects A and C.
Fear conditioning test
The fear conditioning test was carried out at 4 weeks after the brain was injected with virus (Hong et al.,2020).A conditioning chamber (a 30 cm3plastic container) was used for the experiments.After acclimating to the training setting for 3 minutes,mice received 3 minutes of impromptu foot shocks (0.5 mA for 2 seconds at 1 minute intervals).On the second day,freezing time was recorded after placing the mice back in the conditioning room for 3 minutes.The testing chamber was wiped with 70% ethanol between each session.
Western blotting
HEK293 cells (Cat# GDC0067,RRID: CVCL_0045,China Center for Type Culture Collection,Shanghai,China) were transfected with tau-N368 and STAT3 plasmids for 48 hours via Lipofectamine 2000 transfection reagent (Thermo Fisher Scientific,Waltham,MA,USA).HEK293 cell line with multiple neuronal properties,expressing a protein structure closest to its conformationin vivoand with high transfection efficiency.After behavioral experiments,mice were anaesthetized by intraperitoneal injection of 2% sodium pentobarbital at a dose of 45 mg/kg body weight (FWD Chemicals Ltd.,Shanghai,China) and sacrificed using the cervical dislocation method.Mouse brain tissue was lysed in radio immunoprecipitation assay (RIPA) buffer (Beyotime Biotechnology,Shanghai,China) at 4°C for 10 minutes.Next,total cellular proteins were extracted.A nuclear extraction kit (Signosis,San Francisco,CA,USA) was used to isolate cytoplasmic and nuclear proteins,as directed by the manufacturer’s protocol.A cell mitochondria isolation kit (Beyotime Biotechnology) was used for cellular mitochondrial protein analysis.Protein levels were measured using a bicinchoninic acid assay (BCA) Protein Assay Kit (Thermo Fisher Scientific,Waltham,MA,USA).Western blotting was performed using the supernatant,after which the proteins were separated on sodium dodecyl sulfate gels and transferred onto nitrocellulose membranes (Whatman,Little Chalfont,UK).Afterward,the membranes were first blocked with 5% bovine serum albumin (BSA) for 1 hour at room temperature before overnight incubation at 4°C with primary antibodies (Additional Table 1).Thereafter,the samples were incubated with anti-mouse (1:10,000,IRDye 800CW,LI-COR Biosciences,Hessen,Germany,Cat# 926-32210,RRID: AB_621842) or anti-rabbit (1:10,000,IRDye 800CW,LI-COR Biosciences,Cat# 926-32211,RRID: AB_621843) secondary antibody for 1 hour at room temperature.Immunoreactive bands were visualized using the Odyssey Infrared Imaging System (LI-COR Biosciences).Relative optical density was evaluated with ImageJ Software (version 1.8.0;National Institutes of Health,Bethesda,MD,USA) (Schneider et al.,2012).
Electrophysiological analysis
Mice were anesthetized and brain tissue was removed from the skull after behavioral experiments.Using a Leica VT1000S Vibratome (Milton Keynes,UK),horizontal slices of the mouse brain measuring 400 µm in thickness were cut in ice-cold oxygenated artificial cerebrospinal fluid (containing NaCl 126 mM,KCl 3 mM,NaHCO324 mM,NaH2PO41.25 mM,CaCl22 mM,MgCl22 mM,and glucose 10 mM,pH=7.4),saturated with 95% O2 and 5% CO2.After that,the slices were transferred and subjected to incubation with artificial cerebrospinal fluid for 1 hour at room temperature.Slices were maintained flat in artificial cerebrospinal fluid (4 mL/min;32°C),and then placed in an 8×8 array of planar microelectrodes (MED P5455;Alpha MED Sciences,Panasonic,Osaka,Japan).Synaptic responses were evoked on mossy fibers using the MED64 System (Alpha MED Sciences,Panasonic).Excitation of mossy fibers enabled the recording of field excitatory postsynaptic potentials in CA3 of the hippocampus.An evoked field excitatory postsynaptic potential amplitude of 30% of the maximal size was used as the stimulation intensity.For recording long-term potentiation (LTP),high-frequency stimulation (100 Hz,1-second duration) was repeated three times.LTP magnitudes were reported as the mean percentage of baseline field excitatory postsynaptic potential initial slope.
Electrophoresis mobility shift assay
After 48 hours of plasmid transfection in HEK293 cells,nuclear proteins were extracted using a Cytoplasmic Cytosolic Protein Extraction Kit (Signosis).The electrophoresis mobility shift assay (EMSA)-STAT3 analysis was carried out following the guidelines provided by the manufacturer (Signosis).At a temperature of 22°C,a biotinylated oligonucleotide probe containing a STAT3 binding domain was allowed to incubate with the nuclear protein fraction for 30 minutes.After that,the samples were isolated on non-denaturing polyacrylamide gels before being electrophoretically deposited onto nylon membranes by means of membrane transfer methods and electrophoresis.Ultraviolet cross-linking (120,000 J) was used to immobilize the transferred oligonucleotides for 1 minute.Finally,the enhanced chemiluminescence instrument (Clinx,Shanghai,China) was used to develop the blots.During competition assays,unlabeled cold probes that contained STAT3 binding domains were used.
Quantitative reverse transcription-polymerase chain reaction
Mice were anesthetized and brain tissue was removed from the skull after behavioral experiments.Total RNA from hippocampal CA3 regions of mice were extracted with the aid of Trizol reagent (Life Technologies,Carlsbad,CA,USA).The instructions provided by the manufacturer were followed to conduct reverse transcription and quantitative reverse transcriptionpolymerase chain reaction (PCR) (TaKaRa,Dalian,China).The PCR kit included forward and reverse primers (concentration of 0.5 µM),5 µL of SYBR Green PCR master solution,and 0.5 µL of cDNA.A StepOnePlus Real-Time PCR Detection System (Life Technologies) was used for quantitative reverse transcription PCR.The expression level of the gene of interest was normalized to the housekeeping gene,β-actin.Primers used for PCR are provided inAdditional Table 2.
Luciferase reporter assay
For 48 hours,HEK293 cells were transfected with either tau-N368 plasmid or its empty vector control (from Chinese Academy of Sciences,Shenzhen,China) in conjunction with the pSTAT3-Luc reporter (Signosis) construct and pRL-TK (Promega,Madison,WI,USA).Measurement of luciferase activity was carried out following the manufacturer’s guidelines,using a dual-luciferase reporter assay kit (Promega).As a proxy for transfection effectiveness,Renilla luciferase activity (pRL-TK) was used to normalize transcription factor (TF) (firefly luciferase) activity.
Immunofluorescence staining
Mice were anesthetized and perfused after behavioral experiments.The whole brain was removed and then fixed with 4% paraformaldehyde.Successive coronal slices of 30 µm were cut with a frozen slicer (Leica,Wetzlar,Hessen,Germany).After washing three times in phosphate buffer saline (PBS),frozen sections were subjected to incubation at room temperature for 30 minutes with PBS containing 0.5% Triton X-100,before subsequent incubation at room temperature for 30 minutes in blocking buffer (PBS with 5% BSA and 0.2% Triton X-100).Thereafter,brain samples were treated with primary antibodies (Additional Table 1) at 4°C overnight,before incubation with secondary antibodies (Alexa Fluor 488 goat anti-rabbit IgG,1:500,Beyotime Biotechnology,Cat# A0423,RRID: AB_2 335700;Alexa Fluor 488 goat anti-mouse IgG,1:500,Beyotime Biotechnology,Cat# A0428,RRID: AB_2 893435;Alexa Fluor 555 donkey anti-rabbit IgG,1:500,Beyotime Biotechnology,Cat# A0453,RRID: AB_2 890132;Alexa Fluor 555 donkey anti-mouse IgG,1:500,Beyotime Biotechnology,Cat# A0460,RRID: AB_2890133) at 37°C for 1 hour.Finally,4′,6-diamidino-2-phenylindole (DAPI) (1:1000,Beyotime Biotechnology,Cat# C1002) was incubated with the slices at room temperature for 10 minutes.A fluorescent microscope (LSM710,Zeiss,Oberkochen,Germany) was used to observe the images.
Nissl staining
Mice were anesthetized and perfused after behavioral experiments.Frozen portions were placed on slides,allowed to air dry,and stained with Nissl reagent as per the manufacturer’s directions (Beyotime Biotechnology).A brightfield microscope (Nikon,90i,Tokyo,Japan) was used to obtain images.
Golgi staining
Mice were anesthetized and sacrificed after behavioral experiments.The mice were first anesthetized with 2% sodium pentobarbital (FWD Chemicals Ltd.) and the whole brain carefully removed.After dissection,brain tissues were immersed in different impregnation solutions as directed by the manufacturer (FD NeuroTechnologies,Columbia,SC,USA,PK401).Subsequently,the brain was sliced into 90 µm slices and the sections stained with an FD Rapid Golgi Stain Kit.After staining,the sections were submerged in gradient alcohol,and the slices dehydrated using gradient alcohol,and finally cleared with xylene (Sinopharm Chemical Reagent,Shanghai,China).The images were analyzed with an Olympus BX60 (Olympus,Tokyo,Japan).Spines were counted in intact dendritic branches in the CA3 region of the hippocampus.
Lactate dehydrogenase assay
Supernatant was collected from the culture medium of HEK293 cells transfected with tau-N368 and STAT3 plasmids for 48 hours.Lactate dehydrogenase (LDH) levels in the culture medium were quantified to assess cytotoxicity.To measure LDH levels,a LDH cytotoxicity test kit was used as directed by the manufacturer (Beyotime Biotechnology).
Cell counting kit 8 assay
HEK293 cells transfected with tau-N368 and STAT3 plasmids for 48 hours were used as samples.Cell counting kit 8 (Beyotime Biotechnology) was used in accordance with the kit instructions to examine cell viability.
Statistical analysis
No statistical methods were used to determine the sample sizes in advance,although our sample sizes are similar to previousin vivoexperiments (Hong et al.,2020;Wan et al.,2021).A blinded process was used for data collection and analysis.Where the number of samples is small,data are reported as mean ± standard deviation (SD);i.e.,for statistical analysis of western blotting,immunofluorescence,luciferase reporter assay,lactate dehydrogenase assay,cell counting kit 8 assay,Nissl staining,and quantitative reverse transcription PCR.Where the number of such samples is large,data are reported as mean ± standard error of mean (SEM);i.e.,for statistical analysis of behavioral experiments and electrophysiological analysis.When comparing two groups,two-tailed unpaired Student’st-test was used.Additionally,comparative analysis among multiple groups was accomplished using either one-way or two-way analysis of variance with Tukey’s or Bonferroni’spost hoctest.GraphPad Prism 7.00 software (GraphPad Software,San Diego,CA,USA,www.graphpad.com) was used to conduct all analyses of statistical data.The significance criterion was established atP<0.05.
Results
Intracellular tau-N368 accumulation inactivated STAT3
Our earlier research showed that accumulation of htau or P301L-htau reduced STAT3 activity (Hong et al.,2020;Wan et al.,2021).AEP-derived tau fragments (tau-N368) are prone to aggregation and strongly trigger neurodegeneration (Zhang et al.,2014;Wang et al.,2018a).We thus speculated that tau-N368 accumulation may influence the activity of STAT3.On the basis of previous studies,the cleaved tau1-368 band was identified using two distinct tau antibodies (HT7 and tau5) (Additional Figure 2),with tau-N368 expression detected using the anti-HT7 antibody.Consistent with our earlier research (Hong et al.,2020;Wan et al.,2021),after transfection of the tau-N368 plasmid into HEK293 cells (Figure 1AandB),or AAV-eGFPtau-N368 virus injection into the hippocampal CA3 region of C57BL/6 mice (Figure 1CandD),we found that overexpressing tau-N368 increased the levels of STAT3 phosphorylation at Tyr705 (pY705),while total STAT3 levels showed no change (Figure 1AandC).However,the nuclear fraction showed a reduction in both total STAT3 and its phosphorylation at Ser727 (pS727),which positively correlated with the transcriptional activity of STAT3 after it shifted subcellular localization to the nucleus (Figure 1BandD).We used immunofluorescence to show that increased levels of tau-N368 lowered nuclear STAT3 levels (Figure 1E).EMSA using an oligonucleotide probe bearing the STAT3 binding domain found that an increase in tau-N368 levels inhibited the capacity of STAT3 to bind to DNA,and the use of a cold probe disrupted this association (Figure 1F).Furthermore,a TF luciferase experiment confirmed that tau-N368 overexpression attenuated STAT3 activity (Figure 1G).These results demonstrate that an increase in intracellular tau-N368 led to suppression of STAT3 activity.

Figure 2|Overexpression of STAT3 ameliorated tau-N368-induced cognitive deficits.
STAT3 overexpression ameliorates tau-N368-induced memory deficits
As STAT3 overexpression attenuated htau/P301L-tau induced behavior disorders (Hong et al.,2020;Wan et al.,2021),we examined the influence of STAT3 overexpression to gain knowledge about the involvement of STAT3 in tau-N368-induced memory and learning impairments.Twomonth-old C57BL/6 mice received bilateral injections of AAV-STAT3 mixed with AAV-tau-N368 into the CA3 region of the hippocampus.STAT3 overexpression was confirmed after 1 month by western blotting (Figure 2A) and immunofluorescence (Figure 2B).We investigated how activation of STAT3 affected spatial learning and memory using the Morris water maze experiment.On days 4-6,learning ability was hampered due to tau-N368 overexpression,shown by a markedly increased time to locate the hidden platform.Increasing STAT3 expression on days 5 and 6 prevented these tau-N368-induced learning impairments (Figure 2C).In memory testing on day 7,we found that STAT3 and tau-N368 overexpressing mice showed a reduced average latency in arriving at the previous target quadrant,more platform crossings,and remained for a prolonged period in the desired quadrant compared with tau-N368 mice (Figure 2D-G).No remarkable difference was observed in swimming velocity across the groups,indicating that motor impairments did not account for the findings (Figure 2H).Mice were tested for their spatial learning and memory using the Barnes maze,which is a behavioral model based on a dry-land environment (Pitts,2018).In the Barnes maze,tau-N368 mice displayed a significant increase in primary latency and primary errors to entering the escape hole on day 4 of the training phase.Overexpressing STAT3 rescued the cognitive deficits induced by tau-N368 (Figure 3AandB).Throughout the probe phase on day 5,we saw that mice overexpressing STAT3 and tau-N368 more frequently entered the exit quadrant (where the escape box was previously hidden) than tau-N368 mice (Figure 3C).Similarly,overexpressing STAT3 improved tau-N368-induced memory deficits,shown by reduced primary latency and primary errors to enter the escape hole (Figure 3DandE).This enhanced long-term memory was further demonstrated by an increase in the freezing period during the memory test of the fear conditioning test (Figure 3FandG).Furthermore,using a novel object recognition test,mice overexpressing STAT3 and tau-N368 spent more time investigating the new object compared with tau-N368 mice (Figure 3H).Overall,these results suggested that activation of STAT3 ameliorated tau-N368-induced cognitive and memory deficits.
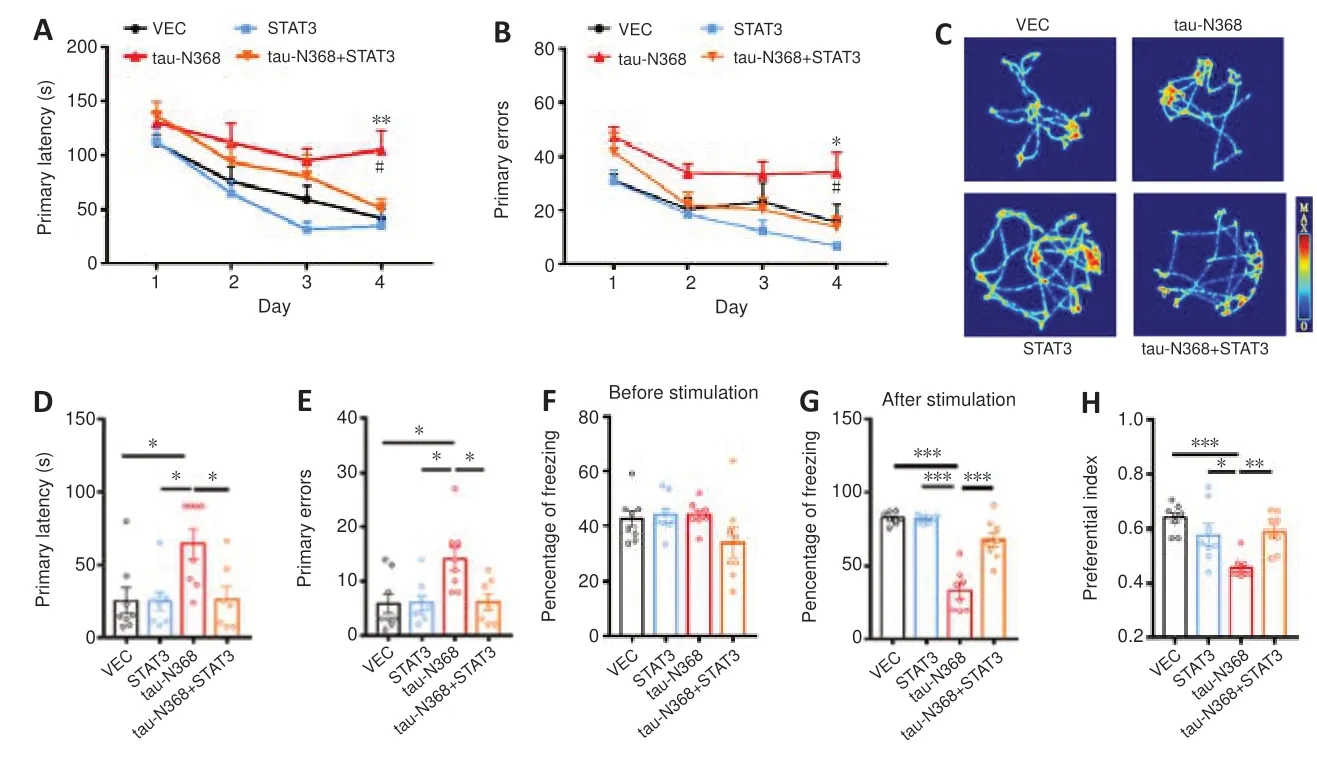
Figure 3|Overexpressing STAT3 ameliorated tau-N368-induced memory deficits.
STAT3 overexpression rescues tau-N368-induced synaptic impairments
The processes of learning and memory are closely linked to synaptic plasticity,with LTP playing a crucial role in both (Shors and Matzel,1997).We found that overexpression of STAT3 considerably mitigated tau-N368-induced suppression of LTP (Figure 4AandB).Golgi staining showed that dendritic spine density was markedly reduced after tau-N368 overexpression compared with controls,but was partially restored by STAT3 overexpression (Figure 4CandD).Our earlier research showed reduced NMDAR protein and mRNA levels with htau or P301L-htau overexpression (Li et al.,2019;Hong et al.,2020;Wan et al.,2021).Therefore,we measured protein levels of NMDARs to determine how an increase in STAT3 levels contributes to tau-N368-induced synaptic impairments.The data demonstrated reduced protein and mRNA levels of GluN2A,GluN2B,and GluN1 due to tau-N368 accumulation.Furthermore,tau-N368-induced downregulation of NMDAR protein and mRNA levels was ameliorated by STAT3 overexpression (Figure 4E-G).These data demonstrated that activation of STAT3 ameliorated tau-N368-induced synaptic plasticity deficits by promoting NMDAR expression.
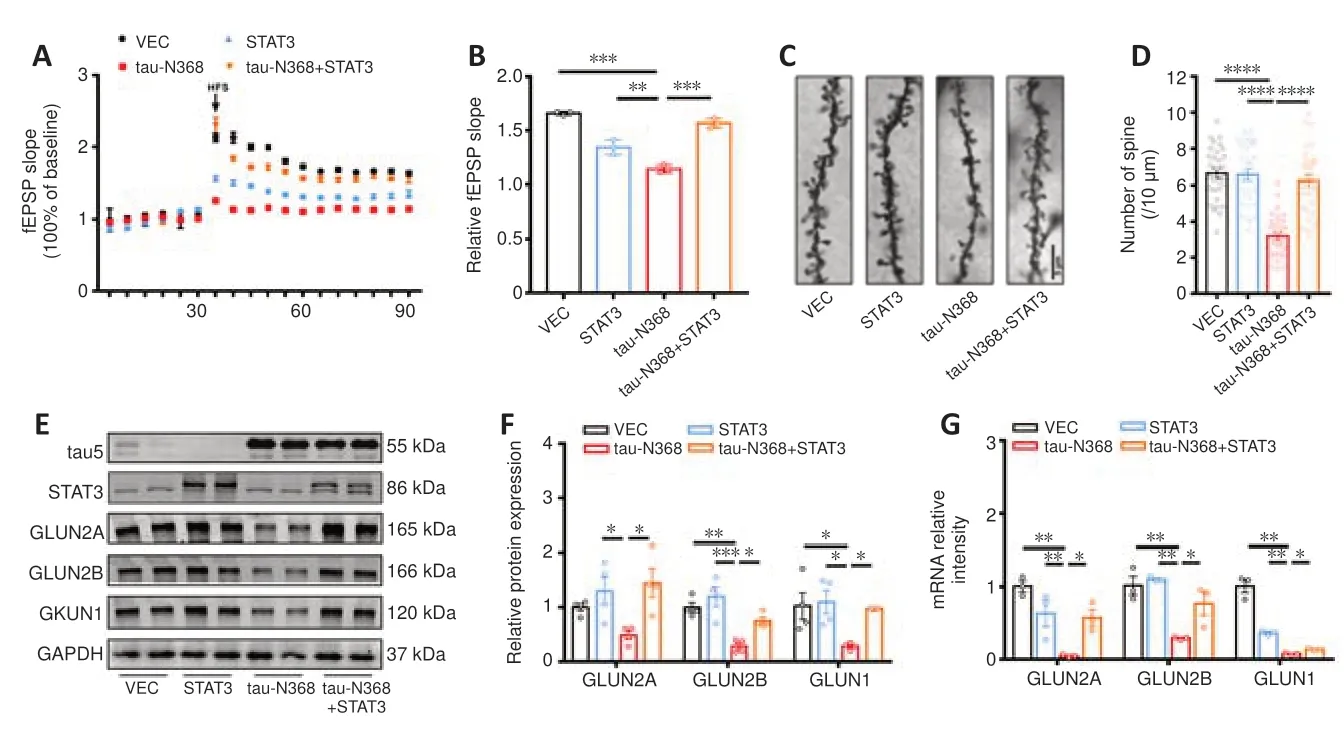
Figure 4|STAT3 Overexpression ameliorated tau-N368-induced synaptic impairments.
STAT3 overexpression attenuates tau-N368-induced cell apoptosis
In contrast with wild-type or other tau fragments,tau-N368 induced substantial apoptosis (Zhang et al.,2014).Using HEK293 cells,we transfected tau-N368 and STAT3 plasmids to determine if STAT3 is implicated in apoptosis triggered by tau-N368.Overexpression of tau-N368 significantly decreased cell survival compared with controls.However,overexpression of STAT3 attenuated tau-N368-induced cell cytotoxicity (Figure 5AandB).Western blotting revealed that STAT3 overexpression reduced the increase of cleaved caspase-3 levels caused by tau-N368in vitro(Figure 5CandD).A common pathogenic phenotype of AD is neuronal death (Deshpande et al.,2019;Jayaraman et al.,2021;Yepes,2021).As expected,STAT3 overexpression reversed tau-N368-induced neuronal loss in the hippocampal CA3 region by immunofluorescence,Nissl staining,and terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay (Additional file 1,Additional Figure 3,andFigure 6A-D).We also found that STAT3 overexpression ameliorated neuronal apoptosis by decreasing Bax protein levels in AAV-tau-N368 mice (Figure 6EandF).We also examined Bax protein levels in the mitochondrial fraction,and found that overexpressing STAT3 reversed the tau-N368-induced increase in total Bax levels in the mitochondrial fraction (Figure 6GandH).

Figure 5|Overexpression of STAT3 ameliorated tau-N368-induced cytotoxicity in vitro.

Figure 6|Overexpression of STAT3 ameliorated tau-N368-induced neuronal loss.
Lastly,we examined the protein levels of total STAT1 and STAT1 phosphorylation at Tyr701 (pY701) in AAV-tau-N368 mice.We found that overexpressing tau-N368 increased the levels of total STAT1 and pY701,while overexpression of STAT3 reversed the increased levels of these proteins induced by tau-N368 (Additional Figure 4).
Discussion
Memory loss and neurodegeneration are positively linked to the abnormal accumulation of tau protein in the brain (Hu et al.,2016;Li et al.,2016;Yin et al.,2016;Feng et al.,2020;Torres et al.,2022;Si et al.,2023).Tau proteolytic fragments can drive neurodegeneration in a fragment-dependent manner through aggregation and transcellular propagation (Quinn et al.,2018).The tau1-368 fragment is produced by δ-secretase cleavage of tau,and promotes neurofibrillary tangle formation,amyloid accumulation,neuroinflammation,and neuronal cell death.This results in significant synapse loss and impairments in both learning and memory abilities (Zhang et al.,2015,2017b,2021;Nizynski et al.,2017;Wang et al.,2017).These studies provide empirical support for the hypothesis that the tau-N368 fragment performs an instrumental function in learning and memory abnormalities,and also provides insight into the mechanism by which tau-N368 fragment exerts its toxicity on the incidence and development of AD.In the current study,we showed that the accumulation of tau-N368 prevented STAT3 from moving to the nucleus,and thus rendered STAT3 inactive.STAT3 overexpression mitigated the synaptic deficits and neuronal loss caused by tau-N368.These discoveries provide light on a previously unknown mechanism responsible for synaptic and memory deficiencies associated with tau-N368 (Figure 7).

Figure 7| Schematic model of STAT3 ameliorated tau-N368-induced toxicity.
The STATs are a range of cytoplasmic TFs responsible for mediating intracellular signaling.They are normally produced by receptors on the cell surface,thereby transmitting effects to the nucleus (Siveen et al.,2014).The seven proteins that make up the STAT family are STAT1,STAT2,STAT3,STAT4,STAT5a,STAT5b,and STAT6 (Kisseleva et al.,2002).STAT protein activation is mediated primarily by the binding of specific phosphorylated tyrosine-and serine-containing peptides in the SH2 or SH3 sites through mitogenactivated protein kinases and Janus kinases.These facilitate phosphorylated STATs to polymerize into dimers and enter the nucleus to bind to target genes and promote their transcription (Shuai and Liu,2003).Here,we found that phosphorylation of STAT3 on Tyr705 was stimulated by tau-N368 in bothin vivoandin vitromodels.However,overexpression of tau-N368 prevented STAT3 from entering the nucleus,as shown by a decrease in STAT3 in the nuclear fraction.These data are similar to those from htau and P301L-tau models (Hong et al.,2020;Wan et al.,2021).The repeat domain of tau protein,namely K18,mediates the acetyltransferase action of tau (Cohen et al.,2013).We speculated that tau-N368,like human total tau and P301Ltau,acetylated STAT1 to bind STAT3 in the cytoplasm,and thus prevent STAT3 transportation into the nucleus (Hong et al.,2020;Wan et al.,2021).Using various techniques such as EMSA,immunofluorescence,luciferase activity assay,and western blotting,we also provide compelling data to demonstrate that STAT3 activity was inhibited by tau-N368 accumulation.Our recent studies found that STAT3 positively regulates NMDAR transcription by directly binding with specific gamma interferon activation site (GAS) elements in the promoters of GluN1,GluN2A,or GluN2B (Li et al.,2019;Hong et al.,2020;Wan et al.,2021).In the present study,tau-N368 inactivated STAT3 to inhibit NMDAR expression,which contributed to synaptic and cognitive deficits.
Tau-N368 has been consistently observed in numerous studies to cause cell apoptosis (Hsu et al.,2014;Zhang et al.,2014;Wang et al.,2018b;Xiang et al.,2019).Tau1-368 fragment exerts its neurotoxic effects in AD by preferentially binding to tropomyosin receptor kinase B receptors,inhibiting neurotrophic signaling and causing apoptosis activation of caspase-3 via inhibition of the phosphatidylinositol 3-kinase/Akt pathway (Xiang et al.,2019).STAT3 is also an important player in carcinogenesis due to its role in regulating cell cycle progression and apoptosis (Subramaniam et al.,2013).Moreover,the loss of STAT3 causes early embryonic lethality (Takeda et al.,1997).Aberrant STAT3 activity contributed to increased levels of anti-apoptotic proteins and cell cycle regulatory proteins (Fletcher et al.,2009;Page et al.,2011).In the present study,we discovered that tau-N368 overexpression reduced cell activity and elicited neuronal death,whereas overexpressing STAT3 rescued tau-N368-induced apoptosis by decreasing Bax/cleaved caspase-3 protein levels.STAT1 was reported to play a critical role in promoting apoptotic cell death,which induces the expression of pro-apoptotic genes,such as caspase-1,Fas,and FasL,and to downregulate the expression of anti-apoptotic genes,including Bcl-2 and Bcl-x (Stephanou et al.,2000,2001).In contrast,related STAT3 family members may antagonize STAT1 and have neuroprotective effects (Zhang et al.,2011).In the present study,we found that both increased STAT1 and decreased STAT3 contributed to neuron loss induced by truncated tau,with the anti-apoptotic effect of STAT3 overexpression partially involved in decreased STAT1 levels and activity,which now awaits further study.
Collectively,our results show that STAT3 translocation from the cytoplasm to the nucleus was inhibited when tau-N368 accumulated within the cell.Cognitive impairment caused by tau-N368 was alleviated by activating STAT3,which successfully reversed synaptic damage and neuronal loss by increasing NMDAR levels and decreasing Bax levels.However,how STAT3 ameliorated tau-N368-induced apoptosis through the mitochondrial pathway remains unclear.This should be further explored in future experimental studies.Furthermore,in the present study we used C57BL/6 mice stereotaxically injected with AAV-tau-N368 which is invasive and has less comprehensive pathology and clinical signs than the tau-N368 transgenic AD model mice.Therefore,future studies with tau-N368 transgenic mice would be more convincing.
Acknowledgments:We thank Prof.Keqiang Ye (Shenzhen University of Technology,Chinese Academy of Sciences) providing plasmid EGFP-tau-N368,Dr.Xiao-Yuan Li (Institute of Biomedical Sciences,Academia Sinica,Taiwan,China) providing the plasmid for wild-type STAT3 (WT-STAT3),and Dr.Fei Liu (Jiangsu Key Laboratory of Neuroregeneration,Nantong,China) providing the plasmid EGFP-N1 encoding the full-length human tau (htau,also termed tau441,tau40,or 2N4R).
Author contributions:Project administration: QF,XH;study design: QF,XH,BZ,HW;experimental implementation: BZ,HW,MM,TL,QL,XW,YH;manuscript draft and revision: GL.All authors read and approved the final version of the manuscript.
Conflicts of interest:The authors declare that they have no conflicts of interest.
Data availability statement:All data generated or analyzed during this study are included in this published article and its Additional files.
Open access statement:This is an open access journal,and articles are distributed under the terms of the Creative Commons AttributionNonCommercial-ShareAlike 4.0 License,which allows others to remix,tweak,and build upon the work non-commercially,as long as appropriate credit is given and the new creations are licensed under the identical terms.
Additional files:
Additional file 1:Terminal deoxynucleotidyl transferase-mediated dUTP nick end-labeling (TUNEL) assay.
Additional Figure 1:Animal experimental flow chart.
Additional Figure 2:The immunoprecipitated bands of tau-N368 wasrecognized by HT7 and tau5.
Additional Figure 3:Overexpression of STAT3 ameliorated tau-N368-induced neuronal loss.
Additional Figure 4:Overexpression of STAT3 decreases the level of STAT1 and pY701.
Additional Table 1:The antibodies used in the study.
Additional Table 2:Primers for quantitative reverse transcription-polymerase chain reaction used in the study.

- 中国神经再生研究(英文版)的其它文章
- Could mammalian inorganic polyphosphate be a crucial signaling molecule in neurological disorders?
- Use of an immunocapture device to detect cytokine release in discrete brain regions
- New immune regulators of sciatic nerve regeneration? Lessons from the neighborhood
- Multifunctional glycolipids as multi-targeting therapeutics for neural regeneration
- Astrocytes dynamically regulate the blood-brain barrier in the healthy brain
- Epigenetic memory of drug exposure history controls neural stem cell quiescence in the adult brain