
甲氨蝶呤对器官毒性的作用机制研究进展
2024-02-04 06:15尹连红彭金咏
中国药理学通报 2024年2期
陈 浩,尹连红,彭金咏
(大连医科大学 1. 附属第一医院、2 药学院,辽宁 大连 116044)
甲氨蝶呤(methotrexate,MTX)为抗叶酸类药物,可抑制细胞增生及诱导细胞凋亡,因而广泛应用于自身免疫性疾病以及恶性肿瘤,是目前最重要的控制性抗风湿药物和广泛应用的抗代谢类肿瘤药之一。MTX属于细胞周期特异性药物,主要作用于细胞周期的S期。MTX通过以下两种途径抑制肿瘤细胞的增殖和生长[1],其与二氢叶酸还原酶有高度亲和力,通过竞争性抑制二氢叶酸还原酶活性阻止二氢叶酸还原为四氢叶酸,减少5,10-甲酰四氢叶酸的产生以干扰DNA的合成;MTX也可以阻止嘌呤核苷酸的合成,从而干扰蛋白质的合成。当MTX大剂量应用时,透过血脑、血睾等屏障的药量较高,故能有效地治疗血运不佳的实体肿瘤,如急性淋巴细胞白血病、非霍奇金淋巴瘤等。但是随着使用剂量的增加,其不良反应的程度也相应增加,例如口腔及消化道黏膜损伤、肝肾功能损伤等。本文旨在综述MTX对各器官毒性的作用机制,为今后药理毒理学理论研究以及临床上中毒后的个体化治疗提供参考。
MTX是一种非选择性叶酸拮抗药,在抑制肿瘤细胞增殖的同时,也会抑制正常细胞的生长,产生不良反应。MTX的浓度与其杀伤程度密切相关,而杀伤程度与其引起的不良反应直接相关。大剂量的MTX可以有效杀伤肿瘤细胞,减弱或解除肿瘤细胞引起的免疫抑制,提高机体免疫功能,但大剂量MTX可在机体蓄积,增加不良反应的风险。剂量越大,患者胃肠道反应、肝功能损伤、黏膜损伤、骨髓抑制等不良反应的发生率增加。小剂量的MTX(≤30 mg/周)用于风湿的治疗,长期使用也会发生不良反应,如肝功能异常、骨髓抑制等(Tab 1)。
1 消化道
1.1 MTX致消化道损害的临床表现恶心、呕吐、食欲不振是MTX最常见的消化道毒性反应,在用药后的1~2 d反应最为剧烈。此外,部分病例还表现出口腔黏膜炎和腹泻,并可出现肛门、直肠黏膜炎以及出血等症状,少数病例会继发严重的感染,甚至危及生命。
1.2 病理、生化改变MTX损害消化道后,会出现肠上皮细胞绒毛的融合和萎缩、刷状缘和表面肠细胞的破坏、有丝分裂减少、隐窝消失、隐窝细胞破坏或扭曲、隐窝脓肿形成、杯状细胞排空增加以及炎症细胞(多核细胞和淋巴细胞)广泛浸润。MTX能引起大鼠肠黏膜炎的组织形态学改变以及血管扩张和黏膜充血水肿;对消化道功能方面的影响包括肠黏膜屏障受损、通透性增加,肠系膜淋巴结细菌培养阳性。在分子水平上,肠黏膜蛋白质、DNA及RNA含量明显下降。Kushwaha等[2]通过使用单细胞凝胶电泳测定技术发现MTX能诱导小鼠多个器官的DNA及RNA含量显著下降。因此,MTX被认为能通过直接抑制上皮细胞DNA的合成或加速小肠隐窝细胞的凋亡从而诱发急性小肠黏膜炎。
1.3 MTX致消化道损害的机制
1.3.1半胱天冬酶 在体内,MTX诱导肠道上皮细胞凋亡、肠道黏膜溃疡,造成营养不良和肠道黏膜屏障破坏,从而增加肠道相关脓毒症的风险。MTX对快速增殖的细胞,包括正常的肠道上皮细胞具有很高毒性,如MTX可诱导RIE-1细胞凋亡,减少细胞数量。Papaconstantinou等[3]建立MTX诱导的肠道细胞凋亡的体外模型,并发现MTX处理RIE-1细胞使其线粒体肿胀和膜碎裂,从受损的线粒体中释放细胞色素C诱导肠上皮细胞凋亡。细胞色素C作为一个位于线粒体膜上的电子传递链中的蛋白质成员,被释放到细胞质中与凋亡蛋白激活因子(Apaf-1)结合并激活半胱天冬酶caspase-9,从而激活半胱天冬酶caspase-2和3导致DNA片段、细胞核凝结以及细胞骨架和结构蛋白的破坏。这表明这些特异性半胱天冬酶可能介导了MTX对肠上皮细胞的凋亡作用。Papaconstantinou等进一步研究确定了被MTX激活的特异性半胱天冬酶,并发现一种通用的半胱天冬酶抑制剂,即Z-VAD-FMK,可以有效抑制MTX诱导的RIE-1细胞凋亡。
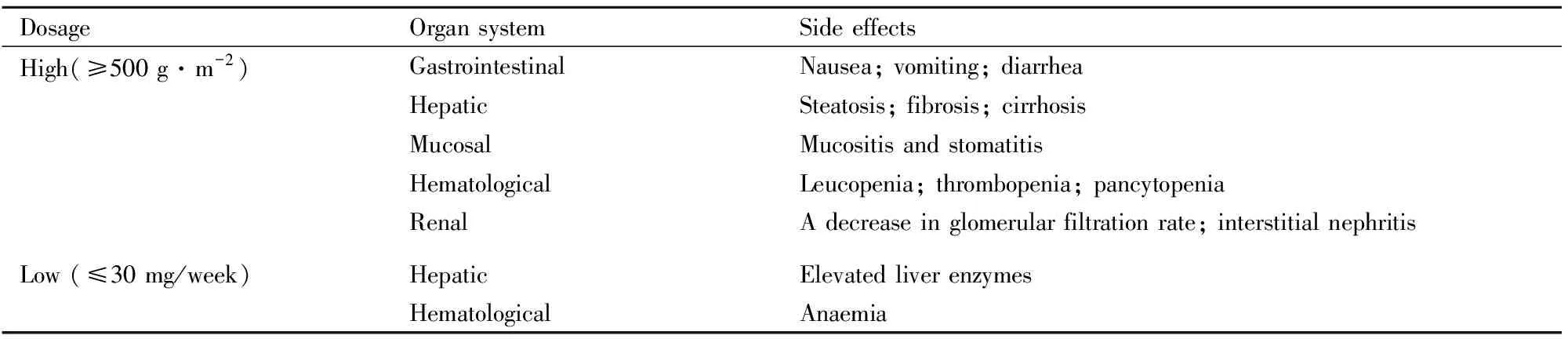
Tab 1 The adverse effects produced by different doses of MTX
综上所述,半胱天冬酶在MTX诱导的RIE-1细胞凋亡中发挥重要作用,是减轻其肠道相关毒性的潜在治疗靶点。
1.3.2二氢叶酸还原酶 MTX致肠黏膜炎的另一种机制是通过抑制肠黏膜层的二氢叶酸还原酶抑制DNA合成,镜下表现为肠黏膜隐窝变浅、绒毛萎缩。MTX使肠黏膜层细胞有丝分裂在隐窝中停止,使覆盖绒毛的上皮细胞减少。微绒毛与脂肪吸收有关,因此脂肪只在MTX处理的大鼠的肠道刷状边界相对少数正常留存的肠道细胞中被吸收。Williams[4]观测到钝化的绒毛可以将其表面积减少到正常值的20%,这表明了小肠的微绒毛吸收能力显著降低。这些改变可能造成消化不良从而引起食欲不振以及体质量减轻。
1.3.3寡肽转运蛋白1 (PEPT1) 载体介导的转运系统在调节药物和营养素的吸收方面发挥着重要作用。PEPT1是载体介导转运系统中一种主要存在于小肠上皮细胞的质子依赖型转运蛋白,主要转运食物中蛋白质水解产生的二肽、三肽。许多研究人员已经证明小肠的病理变化会改变药物吸收,而MTX会诱导肠道药物被动吸收,以及葡萄糖和营养物质的主动吸收,可能是通过干扰PEPT1实现的[5-6]。
氨基-β-内酰胺类抗生素头孢氨苄(cefalexin, CEX)是一种亲水性离子化合物,主要通过活性转运体PEPT1从小肠吸收,而代谢很少。Naruhashi等[7]通过CEX来评估MTX损伤的小肠的吸收功能。CEX总量的变化是活性转运功能的一个指标,并且通过MTX诱导的肠黏膜组织病理学变化可以很好地反映出来。结果发现MTX会影响蛋白质在体内的消化吸收过程,导致恶心、体质量下降、腹泻等临床症状,并可能造成由免疫力低下引发的感染。
1.3.4水、电解质 MTX引起的肠道损伤的特点包括钠、钾分泌增加以及黏膜吸收表面积显著减少。电解质通量紊乱和肠道屏障破坏在很大程度上导致了化疗患者的胃肠道症状(即厌食、恶心、呕吐、腹泻),以及化疗对患者有害的营养影响。Carneiro-Filho等[5]证明了MTX诱导了肠道电解质和水通量的紊乱。对照组Wistar大鼠用MTX治疗3 d以诱发黏膜炎,钠的吸收和钾的分泌明显增加,这些变化与动物出现腹泻相关。
1.3.5溶酶体 多项研究结果显示,MTX处理后的大鼠十二指肠和血浆中瓜氨酸浓度明显偏低[6,8]。在MTX处理的大鼠中,肠细胞合成的这种非蛋白氨基酸的浓度偏低可以反映肠细胞数量的减少,并可能表明吸收功能的降低[9]。这意味着,MTX治疗诱导的黏膜萎缩与瓜氨酸下降相关,而非该药物的厌食症作用;而且十二指肠和血浆中氨基酸谱的紊乱与蛋白质的水解有关。
在大多数细胞中,蛋白质水解由3个主要酶系统的活动产生:溶酶体、Ca2+激活和蛋白酶体途径。组织蛋白酶D(cathepsin D, Cath-D)介导的蛋白水解被认为是溶酶体活性的标志,而在小肠结肠炎的急性期,MTX治疗特异性增强了Cath-D蛋白水解。Leblond等[10]通过实验证实了Cath-D蛋白水解途径的改变,尤其是溶酶体中Cath-D活性增强和蛋白酶体活性减弱。在实验中,MTX处理增加了Cath-D的mRNA表达和活性。其活性的显著增加可能反映了其他溶酶体组织蛋白酶的激活增强,因为组织蛋白酶D激活了其他组织蛋白酶,例如组织蛋白酶B或L。黏膜萎缩和肠道通透性与Cath-D活性相关,这表明了溶酶体途径在MTX诱导的损伤中有潜在作用,而且MTX治疗后空肠中激活的主要蛋白水解途径是组织蛋白酶D系统。
2 中枢神经系统
2.1 MTX致中枢神经系统损害的临床表现大剂量MTX在通过静脉或鞘内注射后出现的中枢神经系统毒性,这是MTX的严重不良反应之一,根据剂量、输液速度、患者自身情况等差异可分别引起急性、亚急性和慢性中毒。急性中枢神经系统毒性通常在给药后数小时内发生,患者可出现类似化脓性脑膜炎的症状,如嗜睡、头痛、恶心呕吐,这些症状一般是一过性的[11-12]。亚急性和慢性中枢毒性可在给药后数日或数周发生,其中患者在亚急性中毒时可能出现类似脑卒中的症状,如半球综合征、失语、构音障碍、吞咽困难和复视等。在慢性中毒过程中,MTX的特异性综合征是坏死性脱髓鞘白质脑病,表现为精神错乱、嗜睡或易激惹、语言障碍等[13],严重病例会出现瘫痪、昏迷甚至死亡,有些患者尤其是儿童可获得部分恢复或病情趋于稳定。
2.2 病理、生化改变MTX急性神经中毒时脑脊液检查可见脑脊液细胞增多、蛋白含量增加、颅内压增高。而亚急性以及慢性神经中毒主要累及深部脑白质尤其是脑室周围和半卵圆中心,从而引起白质脑病,其镜下表现为脱髓鞘、轴突损伤等特征性改变。
2.3 MTX致中枢神经系统损害的机制
2.3.1同型半胱氨酸(Hcy) MTX诱导的神经毒性(MTX-Ntox)的机制有很多,但其中一个潜在生化途径涉及叶酸依赖的Hcy的再甲基化,MTX治疗可增加血浆和脑脊液中的Hcy[14]。Quinn等[15]实验表明,未暴露于MTX的个体的脑脊液Hcy浓度较低,这与之前的少数报道一致[16],而MTX可显著提高脑脊液中Hcy浓度,并且这种作用可能持续至少1周。在脑脊液中,MTX会耗尽细胞内还原叶酸的浓度,从而阻止Hcy向蛋氨酸的再甲基化,这可能在MTX-Ntox的发病机制中起关键作用。
Hcy代谢的一个相关方面也可能是MTX神经毒性的发病机制中的重要组成部分。兴奋性氨基酸(EAAs),如Hcy和半胱氨酸亚磺酸(CSA)以及相关化合物同型半胱氨酸磺酸(HCSA)和半胱氨酸(CA),可能分别从Hcy和CA中氧化衍生出来。这些化合物被认为是神经递质,是NMDA受体的内源性激动剂,是谷氨酸受体的一种亚型。EAAs被认为在癫痫发作的发病机制中起着重要作用。并且可能是典型同型半胱氨酸尿症中癫痫发作的原因。此外,EAAs的过度表达可能通过谷氨酸受体过度激活导致神经元损伤和死亡。这种损伤机制被称为兴奋性毒性,它被认为是许多慢性神经退行性疾病和急性神经病变的共同途径。
Hcy的增加也会导致毒性代谢物S-腺苷同型半胱氨酸(SAH)的病理增加。SAH已被证明是S-腺苷甲硫氨酸(SAM)依赖的甲基转移酶反应的有效产物抑制剂,具有合成5-羟色胺和多巴胺以及维持髓鞘形成所需蛋白质的转甲基作用[17]。SAH可能通过干扰正常内皮细胞中RAS的甲基化,阻断其向细胞膜的移位和RAS功能来抑制细胞生长。
2.3.2半胱天冬酶 曾有病例[18]的研究结果支持氧化磷脂酰胆碱(OxPC)作为急性神经毒性中氧化应激的潜在生物标志。Miketova等[19]在被诊断为低、标准或高风险急性淋巴细胞性白血病(ALL)的21名儿童的脑脊液(CSF)样本中测量出了CNS细胞膜中最常见的磷脂——磷脂酰胆碱(PC)的氧化和未被氧化成分。其中高危儿童在最密集的治疗阶段(巩固期)都接受了最高剂量的MTX治疗。结果表明,对ALL的化疗增加了氧化PC的比例,增加了氧化应激是治疗诱导的中枢神经系统损伤机制的可能性。
Taylor等[18]所报告的3个病例中最一致的生物标志物变化是急性毒性期间caspase-3、7的增加,毒性作用后F2异前列烷水平的增加,以及稍晚发生的氧化磷脂的增加。在急性毒性作用之前和期间,F2异前列烷和caspase-3、7的进行性增加表明MTX诱导氧化应激和凋亡的相关增加。由于PC和PI构成细胞膜,这种氧化PC和PI水平增加的轻微延迟可能反映了氧化应激和凋亡诱导的作用。
2.3.3腺苷 MTX的神经毒性也可能与MTX诱导的腺苷在中枢神经系统的释放和积累有关。MTX及其聚谷氨酸酯的直接抑制作用或继发于DHFR抑制的MTX多谷氨酸累积间接抑制5-氨基咪唑4-甲酰胺核糖核苷酸(AICAR)转化酶引起导致5-氨基咪唑4-甲酰胺核糖核苷酸在细胞内积累,这增加了腺苷的释放和积累。这些腺苷通过作用于穹窿周围外侧下丘脑的A1受体,调节觉醒和嗜睡。这解释了一些患者在摄入MTX后出现的乏力和嗜睡现象。
3 肝脏
3.1 MTX 致肝脏损害的临床表现长期大量MTX治疗会引起血清氨基转移酶升高,从而诱发或加重脂肪肝、肝纤维化甚至肝硬化等疾病。临床上MTX诱发肝损伤的发生率接近70%,并成为MTX临床治疗中给药中断的主要原因。有研究表明MTX在肝脏中抑制RNA和DNA合成并产生细胞停滞从而引起肝损伤的这种机制是直接毒性[20]。还有学者认为MTX导致肝损伤的机制可能是通过选择性干扰肝实质细胞代谢的某一环节,影响肝脏蛋白质的合成[21]。通过近年来研究分析总结后发现MTX致肝损伤机制主要包括叶酸途径、氧化应激途径、腺苷途径和Hcy途径。
3.2 病理、生化改变MTX患者肝活检的组织学变化包括脂肪变性、星状细胞肥大和肝纤维化。
3.3 MTX致肝脏损害的机制
对于事业单位而言,把好人员进口关十分关键,必须不断完善公开招聘、岗位竞聘制度,规范的招聘程序,根据“用进废退”规律可知,人力资源可以通过使用实现增进,闲置只会让人力资源一步步丧失资本存量,同时只有在人力资源开发中引入竞争机制,方可令员工在竞争中不断发展,让有能力、有才华的人脱颖而出。
3.3.1叶酸途径 MTX作为一种二氢叶酸还原酶抑制剂,能够通过抑制二氢叶酸还原为四氢叶酸来减少DNA合成,从而抑制细胞增殖造成肝损伤。同时,位于肝窦周隙(Disse隙)中有一种能储存维生素A的脂肪细胞被称为肝星状细胞(hepatic stellate cell,HSC)。当被MTX诱发的慢性肝损伤激活时,它们会形成骨髓成纤维细胞,分泌胶原蛋白和其他基质蛋白,如纤维连接蛋白。这一过程分为启动和持续两个阶段。在启动阶段,当肝实质细胞受到损伤时,邻近的肝细胞、库普弗细胞等通过旁分泌作用可分泌如肿瘤坏死因子(TNF-α)等多种细胞因子,作用于HSC并使之出现肌成纤维细胞(myofibroblast,MFB)样表型转化,激活并引起细胞增殖、ECM合成增加等。在持续阶段中,细胞因子维持HSC的激活状态并形成纤维。在这一阶段,HSC的活化受自分泌和旁分泌的双重调节。激活的HSC有两个去向:(1)由激活态变回静止态。(2)发生细胞凋亡而死亡。此过程可能解释了MTX诱发肝纤维化以及肝硬化的原因。
3.3.2氧化应激途径 氧化应激是指体内氧化与抗氧化作用失衡的一种状态,是与体内自由基和其他分子化学反应有关的细胞损坏。此时在机体的有氧代谢过程中,线粒体产生的ROS自由基处于高水平状态。研究结果表明,MTX通过增加肝组织中的脂质过氧化和降低抗氧化酶的水平而引起氧化组织损伤。其中MTX产生ROS的一种机制涉及四氢生物蝶呤,它是许多酶的重要辅助因子,也是内皮型一氧化氮合酶(eNOS)的配体,可通过氧化的二氢生物蝶呤转化。四氢生物喋呤促进一氧化氮的生成,而二氢生物喋呤解偶联eNOS,导致产生超氧化物[22]。MTX诱导产生过量的ROS后,会通过如下通路诱导细胞凋亡:
(1)Fas通路:ROS通过Fas/FasL激活半胱天冬酶中的caspase-8,促进细胞色素C的释放,导致caspase-9和3的激活,最终诱导细胞凋亡。
(2)P53/MPTP通路:过量的ROS可以调控P53/线粒体膜通透性转换孔(P53 /MPTP)通路。转录因子P53家族受DNA损伤的活化后能够诱导Bax(一种Bcl-2基因家族中细胞凋亡促进基因)的转录,最终诱导细胞凋亡。
(3)NF-κB通路:ROS可促进蛋白激酶C (PKC)的激活,通过降解IKBs(NF-κB抑制蛋白)的方式活化核转录因子NF-κB,使其进入细胞核内与相应DNA结合,促进目的基因转录,诱导细胞凋亡。
(4)JNK通路:JNK是丝裂原活化蛋白激酶(MAPK)家族中的一员,参与多种应激反应。细胞因子刺激和环境应激均会激活细胞内的JNK。在特异性刺激后,JNK可以被MAPK家族中的2种蛋白激酶MKK4和MKK7激活,并使细胞质中的Bax二聚体或者Bax磷酸化,从而向线粒体移位,诱导线粒体内膜释放细胞色素C,激活caspase-9和caspase-3;也可以使抗凋亡蛋白Bcl-2和Bcl-xl磷酸化,这些都诱导了凋亡发生。
3.3.3腺苷途径 MTX进入机体后部分经肝细胞代谢转化为多谷氨酸盐集聚在肝脏等组织器官中。MTX多谷氨酸盐能阻断将AICAR转化为5-甲酰胺基咪唑-4-甲酰胺核苷酸(FAICAR)的ATIC[23]。已知AICAR可以抑制腺苷脱氨酶,从而防止腺苷分解为肌苷。因此当AICAR水平升高时,细胞内腺苷水平也升高。腺苷是一种旁分泌信号分子,可以结合4种不同的G蛋白偶联受体,分别为腺苷受体A1、腺苷受体A2a、腺苷受体A2b和腺苷受体A3。其中,A1和A2a受体对腺苷具有高亲和力,而A2b和A3受体则相反。目前认为通过腺苷信号传导观察到的大部分临床效应是通过A2a受体。Chan等[23]研究表明腺苷A2a受体刺激肝星状细胞产生胶原。
3.3.4Hcy途径 MTX也可以通过干扰Hcy产生甲硫氨酸的能力来引起肝纤维化。过量的Hcy诱导内质网应激,促进肝脏中脂肪的积累。Hcy还能激活HSC和促炎细胞因子,导致肝纤维化[11,24]。
4 肾脏
4.2 病理、生化改变持续注射一定剂量MTX后的12 h内,在肾皮质观察到出现大面积严重的肾小球和肾小管肾炎,如纤维组织严重浸润、肾小球轮廓破坏或坏死、肾小囊或间隙消失。在上皮层褶皱中可以看到肾小管坏死,小管周围可见纤维沉积,髓质内可见轻度肾小管水肿和扩张。
4.3 MTX致肾脏损伤的机制
4.3.1过敏反应 高浓度MTX可使血清白蛋白变性,使其成为具有免疫原性的载体蛋白。同时,两者可因MTX的半抗原性而结合,并在结合后使人体产生针对MTX-白蛋白复合物的抗体[25],从而引起超敏反应,通常表现为间质性肾炎。间质性肾炎所导致的肾小管功能障碍可出现低比重及低渗透压尿、肾小管性蛋白尿和水、电解质、酸碱平衡紊乱等。
4.3.2氧化应激 谷胱甘肽(GSH)是一种重要的细胞溶质抗氧化剂,对ROS具有保护作用。在正常条件下,谷胱甘肽还原酶利用还原型辅酶(NADPH)维持细胞内GSH的还原状态。MTX可抑制细胞溶质烟碱依赖腺苷二磷酸(NADP)的脱氢酶类和NADP苹果酸酶(NADP-ME),从而降低细胞内NADPH的可用性。MTX使GSH水平的显著降低导致抗氧化酶防御系统的有效性降低,使细胞对ROS敏感。而且有学者研究发现,在大鼠模型中长时间低剂量MTX给药会导致严重的肾损伤和肾MTX蓄积,且比短时间给药更加严重[26]。
4.3.3MTX代谢产物积聚抑制正常代谢 MTX在体内有部分通过胃肠道细菌代谢,而更多经肝细胞代谢转化为谷氨酸盐。超过90%的MTX以原形被肾脏清除,而少量MTX及其代谢产物能够贮存于肾脏和肝脏等组织中长达数月。经肾小球滤过及肾小管分泌而形成的代谢产物7-羟基MTX溶解度较低,易形成结晶。尤其在酸性环境中,此时代谢产物7-羟基MTX和丹皮酸的溶解度比MTX低6~10倍。该药及其代谢产物的浓度超过2×103mol·L-1时在酸性尿中(pH<5.5)沉积,尿液的pH从6.0增加到7.0会使其溶解度增加5到8倍,而在pH>7时其溶解度可增加10倍。在高剂量MTX治疗时,尿液中MTX及其代谢产物浓度容易超饱和,从而产生结晶堵塞肾小管,长时间堵塞肾小管诱发肾功能障碍从而导致MTX消除延迟,由此引起的持续升高的血浆MTX浓度可能造成急性左心室衰竭和MTX其他毒性的显著增强。而结晶破坏崩解产生大量尿酸经肾脏排泄,在肾小管沉积引起肾小管功能损害,临床上表现为血尿、蛋白尿、尿少、氮质血症、尿毒症等。
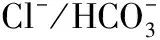
虽然体内的细胞死亡会导致间质炎症,然而临床研究显示只有少量或没有肾小管毒性和炎症反应的迹象。因此可以推测,容量调节机制在影响MTX诱导的细胞肿胀后大量细胞死亡方面更重要。同时实验发现长期暴露于MTX后细胞死亡显著增加,这表明如果MTX消除延迟,患者肾损害的风险增加。
4.3.5基因多态性 研究发现MTX的肾脏毒性与亚甲基四氢叶酸还原酶(MTHFR)的基因多态性有关,而且在临床上相同剂量MTX治疗不同个体时不良反应轻重不同。MTHFR是人体叶酸代谢中的一个重要的酶,是叶酸代谢的核心。它能够通过催化5,10-亚甲基四氢叶酸转换成5-甲基四氢叶酸盐从而为Hcy提供甲基形成甲硫氨酸。MTX通过阻断MTHFR将5,10-亚甲基四氢叶酸转化为5-甲基四氢叶酸来抑制DNA复制。而MTHFR具有两种常见的功能多态性:C677T和A1298C。Suthandiram等[28]对马来西亚71例接受MTX治疗的成人血液恶性肿瘤患者的MTHFR C677T和A1298C基因多态性与MTX不良反应相关性进行的研究结果显示,MHFR C677T和ABCB1 C3435T多态性似乎与更高的MTX血浆浓度相关(P<0.05)。C677T基因型被认为与MTX肾脏毒性或骨髓抑制相关,目前并无定论。
5 肺
5.1 MTX致肺损伤的临床表现MTX能引起包括肺实质炎症、肺间质炎症、气道高反应、肺癌等多种肺损伤。临床表现包括急性发作性呼吸困难;呼吸急促频率>28次/min,伴干咳;发热>38.0 ℃,大多数患者查体可闻及肺底部啰音[29]。
5.2 病理、生化改变肺部影像学表现常为弥漫性斑片状双侧磨玻璃影,病理检查提示弥漫性肺泡损伤和局灶性肺纤维化。
5.3MTX致肺损伤的机制
5.3.1直接细胞毒性 MTX可诱导肺泡上皮细胞损伤和凋亡,导致肺泡毛细血管屏障基底膜的完整性丧失。随后肌成纤维细胞积聚、增殖,胶原沉积。
5.3.2细胞因子和p38丝裂原活化蛋白激酶(MAPK)通路 p38 MAPK信号通路能被炎性细胞因子和环境应激激活,从而引起自身免疫性疾病。在三种主要的MAPK途径中,p38和JNK途径与慢性炎症有关。促炎和抗炎细胞因子是肺部炎症中的重要介质。其中抗炎细胞因子包括IL-4、IL-10和TGF-β,而IL-1β、TNF-α、IFN-γ、IL-2、IL-6等是促炎因子。
MTX作为一种临床免疫抑制剂,其免疫调节作用基于促炎和抗炎细胞因子水平之间的平衡,其中部分作用是能够抑制纯化T细胞中细胞因子如TNF-α、IFN-γ和GM-CSF的产生。而且在Kim等[30]的研究中,MTX激活IL-1β的表达,并依次诱导p38 MAPK信号蛋白的磷酸化,包括TAK1、MKK3/MKK6、p38 MAPK、MAPK k2和HSP27。此外,HSP27激活可增加IL-8分泌,导致肺部炎症反应。
综上,MTX刺激了p38 MAPK信号级联的成分(TAK1 → MKK3/MKK6 → p38 MAPK → MAPKK2 → HSP27)并促进IL-1β和IL-8的释放。此外,p38 MAPK介导的抗炎细胞因子IL-4途径的抑制可能在MTX诱导的肺部炎症中发挥作用。
6 讨论
由于MTX对各器官毒性机制有所差异,而且个体差异性较大,因此可在临床上将MTX与其他药物联合应用,以减少副作用的发生。例如在治疗过程中密切监测其血药浓度,根据其变化及时调整四氢叶酸钙(CF)解救方案,以期达到治疗有效性的同时保证用药安全性。同时也要视情况进行充分的水化碱化治疗、保肝治疗、必要时给予输注成分血制品以及感染发生时积极应用抗生素治疗。现有多项研究表明多种中药成分对MTX不良反应具有一定的治疗效果。因此,完善MTX毒性作用机制的总结,为其未来在临床的合理用药及与不同药物的联用提供新的思路与方向。
猜你喜欢
检察风云(2022年5期)2022-04-05
中国生殖健康(2020年6期)2020-02-01
小哥白尼(野生动物)(2019年5期)2019-08-27
中国生殖健康(2019年12期)2019-01-07
中国生殖健康(2018年6期)2018-11-06
妈妈宝宝(2017年4期)2017-02-25
国外医药(抗生素分册)(2016年3期)2016-07-12
山东医药(2015年16期)2016-01-12
吉林大学学报(医学版)(2015年4期)2015-12-17
发明与创新(2015年33期)2015-02-27
