
铁死亡途径及其相关分子泛素化修饰的研究进展
2024-02-04 06:15杨晓妍周媛静罗秀菊
中国药理学通报 2024年2期
杨晓妍,周媛静,罗秀菊,彭 军
(1.中南大学湘雅药学院药理学系,湖南 长沙 410078; 2. 中南大学湘雅三医院检验科,湖南 长沙 410013)
在疾病发生发展过程中,会发生不同类型的细胞死亡,常见细胞死亡方式有凋亡、坏死和自噬性死亡等,其中凋亡和自噬性死亡受到严密的信号通路调控,而坏死通常是被动和意外的死亡方式,不受信号通路调控。近年来研究发现,某些坏死也受到特定的信号通路调控,称为调节性坏死,包括坏死样凋亡(necroptosis)、铁死亡(ferroptosis)、细胞焦亡(pyroptosis)、PARP-1依赖性细胞坏死(parthanatos)、CypD依赖性坏死等[1]。其中,铁死亡是细胞膜上多不饱和脂肪酸磷脂过氧化损伤引起的一种铁依赖性细胞死亡方式,形态学特征表现为线粒体膜密度增加,线粒体嵴减少或者消失,线粒体外膜破裂,但细胞核正常;生化特征则表现为铁积累和脂质过氧化。铁死亡参与了多种疾病的病理过程,如心梗、脑卒中、癌症和退行性疾病等。
泛素(ubiquitin,Ub)是一种进化上保守的蛋白质,可对蛋白质进行翻译后标记以降解。泛素化修饰(ubiquitination)与细胞内生理和病理过程密切相关,包括细胞周期控制、酶激活、信号转导、转录、DNA修复、受体转运、免疫反应、炎性反应和凋亡等。研究表明,泛素化修饰涉及细胞铁死亡过程,该文将就铁死亡途径及其相关分子泛素化修饰研究进展作一综述。
1 铁代谢简介
铁离子是维持机体功能正常运转必不可少的金属元素,参与构成血红蛋白、肌红蛋白及多种酶,影响氧气运输、能量代谢和信号传递等多种生理过程。铁通常以Fe3+和Fe2+形式存在,Fe3+在十二指肠及空肠上段被铁还原酶还原成Fe2+,随后经肠道吸收或红细胞降解后被铜蓝蛋白氧化生成Fe3+,再与血浆中的转铁蛋白(transferrin,TF)结合,形成TF-Fe3+复合物。饱和的TF与细胞膜上的转铁蛋白受体(transferrin receptor,TFR)有高度亲和力,复合物通过内吞作用进入细胞,质子泵调节内体pH至 5.5,使转铁蛋白释放Fe3+,TF仍与TFR结合。铁还原酶将Fe3+还原为Fe2+,随后通过金属转运蛋白(divalent metal transporter 1,DMT1)转运至胞质,Fe2+由线粒体铁蛋白(ferritin,FN)引导到线粒体进行代谢利用。过量的铁则以惰性铁(即Fe3+)的形式储存在FN中,或者以Fe2+的形式储存在不稳定铁池(labile iron pool,LIP)中。铁释放后,转运蛋白与TFR的亲和力下降约500倍,导致其解离,转运蛋白回到血流中重新捕获Fe3+。铁从内皮细胞(也包括神经元或星形胶质细胞)释放到脑间质的过程很可能涉及FN与铁氧化酶活性的结合,类似于肠上皮中的基底外侧铁转运机制。铁主要由铁输出蛋白(ferroportin, FPN)排出到细胞外[2]。
2 铁死亡途径
2.1 铁稳态调节途径铁离子缺乏会引起贫血等疾病,而铁离子浓度过高即铁超载则会对细胞造成损伤,过量的Fe2+能通过芬顿反应产生活性氧(reactive oxygen species,ROS)导致脂质过氧化。与铁摄取、储存及输出有关的蛋白出现异常均可引起铁稳态失衡,诱导细胞铁死亡。
细胞内的TFR、FPN及FN都受到铁调节蛋白(iron regulatory protein,IRP)的转录后调节,IRP通过与mRNA上的铁反应元件(iron response element,IRE)结合或解离来调节蛋白的水平高低。编码TFR的mRNA在其非翻译区的3’端有多个IRE,FNmRNA和FPNmRNA在其5’端有一个IRE,当IRP与3’端IRE结合时,能促进mRNA表达,而当与5’端IRE结合时,则能抑制mRNA表达。此外,IRP与IRE的结合力高低与细胞内铁含量有关,当铁含量较低时,IRP以高亲和力与IRE结合,稳定了TFRmRNA的表达,但抑制了FPN和FN的mRNA表达,使得TFR水平上调,铁输出与铁储存减少,增加细胞内铁离子含量;相反,当细胞内铁含量较高时,IRP无法与IRE结合,导致TFRmRNA下调,且促进FPN及FN mRNA翻译,减少铁吸收,促进铁输出与过量的铁储存,降低铁含量[3]。
Feng等[4]发现TFR在细胞表面的累积是铁死亡的一个显著特征;我们最近报道,在缺血/再灌注处理后的大鼠心脏以及缺氧/复氧处理后的H9c2细胞中,TFR上调引起铁过载,从而诱导铁死亡[5]。
FN由铁蛋白重链(ferritin heavy chain,FTH)和铁蛋白轻链(ferritin light chain,FTL)组成,FTH包含铁氧化酶中心,将Fe2+氧化为Fe3+,FTL不具有催化活性,不同细胞类型中这两种亚基比例不同,对铁的储存起关键作用。此外,Gao等[6]发现,erastin诱导的铁死亡细胞中核受体共激活因子(nuclear receptor coactivator 4,NCOA4)介导的自噬途径激活,使FN降解,导致其中储存的铁释放到细胞中的不稳定铁池,细胞ROS积累,导致铁死亡。铁输出主要依靠FPN,有研究发现敲除FPN能加速erastin诱导的SH-SY5Y细胞和NESCs细胞铁死亡[7]。
2.3 VDAC途径线粒体VDAC属于真核生物线粒体孔蛋白,是转运离子和代谢物的跨膜通道,介导代谢物通过线粒体外膜运输,VDAC有3种亚型,即VDAC1、VDAC2和VDAC3。铁死亡诱导剂erastin可以通过直接结合VDAC2/3来改变线粒体外膜的通透性,从而降低NADH的氧化速率,诱导铁死亡,因此VDAC对铁死亡有着重要的调节作用。VDAC能被微管蛋白阻断然后关闭,限制呼吸底物进入线粒体,这对癌细胞来说有利于其糖酵解,被称为Warburg效应。进一步研究发现,erastin可以逆转微管蛋白对VDAC的抑制,使VDAC打开,增加其电导率和促进代谢物进出线粒体[10],导致线粒体功能障碍,释放大量的氧化物,最终诱导细胞铁死亡。

2.5 多不饱和磷酸酯的合成及过氧化途径游离多不饱和脂肪酸(polyunsaturated fatty acid,PUFA)通过酰基辅酶A合成酶长链家族成员4(acyl-CoA synthetase long-chain family member 4,ACSL4)与辅酶A连接生成对应的酰基辅酶A衍生物,PUFA-CoA通过溶血磷脂酰胆碱转移酶3(lysophosphatidylcholine acyltransferase 3,LPCAT3)在磷脂中再酯化,生成多不饱和磷酸酯PUFA-PLs,因此,ACSL4和LPCAT3能增强多不饱和磷酸酯的生成,促进铁死亡。PUFA中存在双烯丙基,很容易发生过氧化,所以它们在PUFA中的数量是决定脂质氧化性的主要因素。脂质过氧化物若不及时转化为脂质氢过氧化物并还原为无毒的醇,自由基介导的反应的扩散会导致大量次级产物的生成,破坏膜的完整性,最终导致铁死亡。值得一提的是,ACSL4促铁死亡的作用与GPX4抗铁死亡的作用呈负相关。GPX4表达和/或活性降低,则ACSL4水平升高,细胞铁死亡增强;而GPX4表达和/或活性升高,则ACSL4水平下降,细胞铁死亡减少[12]。见Fig 1。
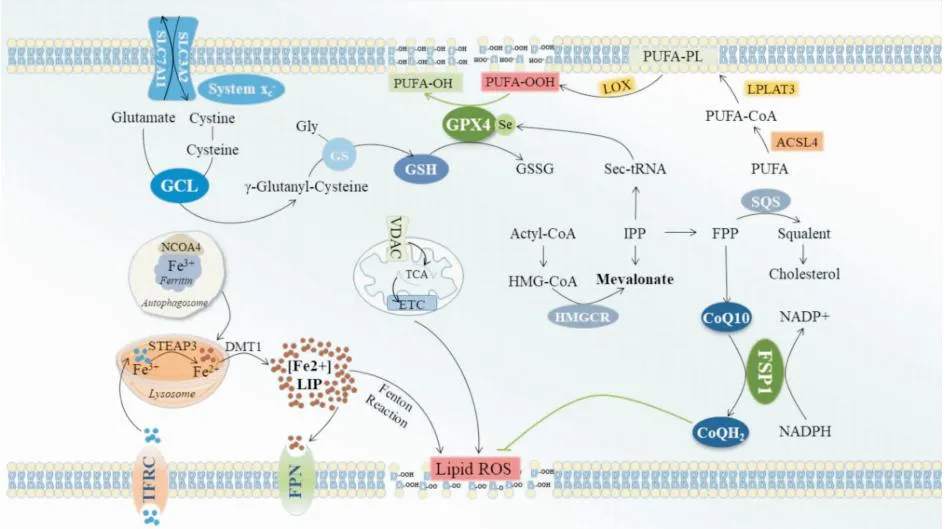
Fig 1 Ferroptosis pathways
3 铁死亡途径相关分子蛋白的泛素化修饰
3.1 转铁蛋白受体的泛素化TFR在维持细胞内铁稳态中发挥重要作用,其蛋白的稳定性将影响到细胞内铁离子水平,进而影响铁死亡。Tribbles同源物2(tribbles homolog 2,TRIB2)可通过调节TFR水平而改变细胞对铁死亡的敏感性,该作用涉及对TFR泛素水平的调节。含有E3泛素蛋白连接酶的β-转导蛋白重复序列(β-transducin repeat containing E3 ubiquitin protein ligase,β-TrCP)是转铁蛋白受体泛素化E3泛素连接酶。一旦β-TrCP被敲除,TRIB2无法降低不稳定铁水平,敲除和过度表达TRIB2对铁死亡和脂质过氧化的影响都离不开TFR和β-TrCP[13]。此外,HECT型E3泛素连接酶(HECT, UBA and WWE domain containing E3 ubiquitin protein ligase 1,HUWE1)可介导肝细胞中的TFR泛素化,降低TFR蛋白水平,阻止细胞内铁超载,进而抑制铁死亡[14]。见Tab 1。
TFR还会受到上游分子泛素化的间接调控。在大鼠心肌缺血/再灌注(ischemia/reperfusion,I/R)模型中,我们课题组发现心肌组织中泛素特异性蛋白酶7(ubiquitin specific protease 7,USP7)、p53和TFR上调,伴随心肌细胞铁死亡增加。USP7抑制剂通过增强p53泛素化,降低p53和TFR水平,减少铁死亡而减轻心肌I/R损伤[5]。Song等[15]将OTU去泛素化酶1(OTU Deubiquitinase 1,OTUD1)鉴定为IRP2的去泛素化酶,在结直肠癌中被选择性还原。OTUD1通过去泛素化和稳定IRP2促进TFR介导的铁转运,导致细胞内铁超载而诱导铁死亡。
3.2 SLC7A11的泛素化OTU去泛素化酶泛素醛结合物1(OTU deubiquitinase,ubiquitin aldehyde binding 1,OTUB1)为调节SLC7A11稳定性的关键因素。OTUB1直接与SLC7A11相互作用并使其稳定;敲低OTUB1则降低癌细胞中SLC7A11水平,抑制小鼠肿瘤异种移植物的生长,这与激活铁死亡有关。过度表达癌干细胞标志物CD44可促进SLC7A11和OTUB1之间的相互作用,增强SLC7A11稳定性;下调CD44能部分减少这种相互作用。总之,CD44以OTUB1依赖性方式抑制癌细胞铁死亡,其在调节SLC7A11的稳定性和癌细胞铁死亡中起着至关重要的作用,是癌症治疗的潜在靶点[16]。
肿瘤抑制剂BRCA1相关蛋白1(BRCA1 associated protein 1,BAP1)编码一种核去泛素化酶,以减少染色质上的组蛋白2A泛素化(H2Aub)。BAP1可降低SLC7A11启动子上H2Aub的占位,并以去泛素依赖性方式抑制SLC7A11的功能,从而抑制胱氨酸摄取,导致脂质过氧化和铁死亡。虽然BAP1能降低SLC7A11启动子上H2Aub部位的结合能力, H2Aub泛素连接酶PRC1却能增强H2Aub结合,但两者都抑制SLC7A11的表达,表明H2Aub的动态调节对SLC7A11的功能至关重要[17]。
Chen等[18]研究了筋膜肌动蛋白捆绑蛋白1(fascin actin-bundling protein 1,FSCN1)在乳腺癌中的表达谱和生物学效应,通过对TCGA癌症数据库的生物信息学分析以及功能获得和缺失的研究表明,Fascin可增强erastin诱导铁死亡的敏感性。Fascin直接与SLC7A11相互作用,并通过泛素介导的蛋白酶体降解途径降低其稳定性。
3.3 GPX4的泛素化倍半萜内酯衍生物DMOCPTL可通过影响GPX4泛素化水平诱导三阴性乳腺癌细胞铁死亡及凋亡,从而抑制癌细胞的增殖,该报道首次证明GPX4泛素化参与调节铁死亡[19]。在高糖诱导人视网膜毛细血管内皮细胞的铁死亡模型,过表达三结构域蛋白质46(tripartite motif containing 46,TRIM46)可增加高糖诱导细胞铁死亡的敏感性,而铁死亡诱导剂RSL3能逆转沉默TRIM46对高糖诱导铁死亡的保护作用。进一步研究发现,TRIM46可以与GPX4相互作用,促进GPX4泛素化[20]。
3.4 VDAC2/3的泛素化神经前体细胞表达发育下调4(NEDD4 E3 ubiquitin protein ligase,Nedd4)是真核生物HECT结构域E3连接酶家族的重要成员,它由1个催化C端HECT结构域、1个N端钙/脂质结合结构域(C2结构域)和4个负责细胞定位和底物识别的WW结构域组成。Nedd4利用其WW结构域识别具有PPxY序列的底物,通过蛋白酶体将其降解。Yang等[21]认为VDAC2/3为erastin诱导的铁死亡中E3连接酶Nedd4的底物,敲低Nedd4导致VDAC2/3蛋白水平升高,使黑色素瘤细胞在体外和体内对erastin诱导的铁死亡敏感性增加。
3.5 NCOA4的泛素化NCOA4为铁蛋白自噬降解的运货受体,它的丰度取决于细胞内铁含量以及与E3泛素连接酶HERC2的相互作用。HERC2仅在铁水平高时与NCOA4结合,使NCOA4被蛋白酶体分解。当铁水平低时,HERC2不与NCOA4相互作用。NCOA4介导的铁蛋白自噬称为铁自噬,参与细胞内铁水平和铁死亡调节。三结构域蛋白质7(tripartite motif containing 7,TRIM7)属于E3泛素连接酶,介导内源性蛋白质被泛素蛋白酶体系统降解。TRIM7通过其C端PRY/SPRY结构域,使用K48连接链直接结合,升高NCOA4泛素化水平,降低NCOA4蛋白水平,进而抑制铁蛋白降解,降低细胞内铁水平,减少细胞铁死亡[22]。
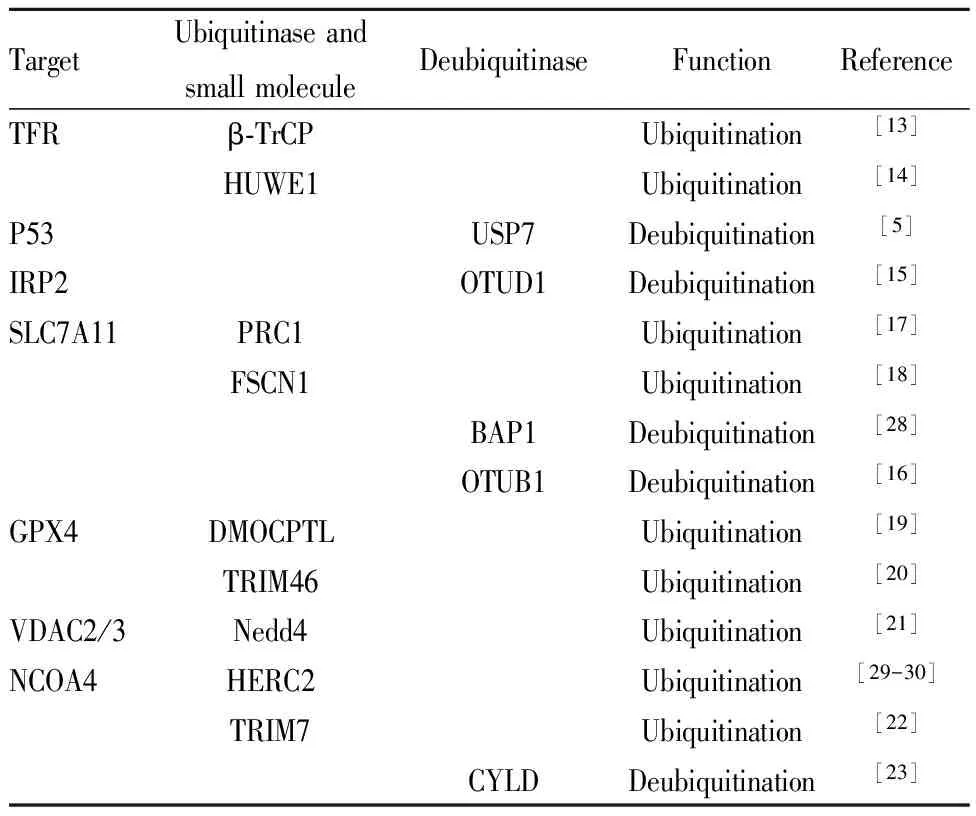
Tab 1 Modification of ubiquitination and deubiquitinationof ferroptosis-related molecules
除TRIM7外,NCOA4还受到多种去泛素化酶的调节,如去泛素化酶圆柱状蛋白(CYLD lysine 63 deubiquitinase,CYLD)。利用小鼠阿霉素心脏毒性模型,我们课题组探讨了精子发生相关蛋白2(spermatogenesis associated 2,SPATA2)/CYLD通路与NCOA4介导的铁自噬之间的关系。我们发现阿霉素处理后,心肌组织SPATA2和CYLD蛋白水平升高,二者相互作用增强,NCOA4泛素化水平降低,NCOA4稳定性增加,导致铁自噬增强,促进了心肌细胞铁死亡和心肌损伤[23]。
4 靶向调节铁死亡途径相关蛋白泛素化水平与疾病治疗
铁死亡涉及多条相关途径,靶向各条途径的铁死亡诱导剂或抑制剂均具有潜在的临床价值。研究表明,通过诱导癌细胞铁死亡来抑制肿瘤生长是一种有效的癌症治疗策略[24],但由于存在特异性低、半衰期短或长期毒性等原因,目前大多数铁死亡诱导剂用于临床还有很长的路要走。由于泛素化修饰系统可调节癌细胞对铁死亡的敏感性,靶向泛素化修饰很可能成为诱导癌细胞铁死亡的新策略。如前所述,诸多铁死亡途径相关分子(如TFR、SLC7A11、GPX4和VDAC2/3等)的稳定性均与其泛素化水平密切相关。理论上,通过靶向干预上述铁死亡相关分子特定的泛素或去泛素酶均可影响铁死亡,进而达到治疗某些疾病的目的。目前这方面的报道不多,主要集中在靶向调节GPX4泛素化水平。
GPX4具有抗细胞铁死亡作用,是铁死亡的标志物之一。靶向调节GPX4的泛素化水平进而促进细胞铁死亡是目前治疗癌症的新策略。20S蛋白酶体抑制剂PIs硼替佐米和卡非佐米已被FDA批准治疗血液系统恶性肿瘤,靶向作用于蛋白酶体系统(ubiquitin-proteasome system,UPS)。由于许多患者对PIs具有耐药性,学者将目光聚焦到了UPS的上游去泛素化酶(deubiquitinase,DUB)上,Yang等[25]发现了一种金属基化合物即钯-吡啶硫酮复合物PdPT,可以通过抑制DUB,促进铁死亡标志物GPX4的蛋白酶体降解,从而促进铁死亡。
蛋白质水解靶向嵌合体(PROTACs)可以通过调节E3泛素连接酶的活性从而引起目的蛋白的蛋白酶体降解。Luo等[26]基于PROTAC策略设计了一种用于GPX4化学降解的小分子dGPX4,它将GPX4抑制剂ML162与可招募E3泛素连接酶的泊马度胺共价结合而成。dGPX4能够在不同类型癌细胞中降解GPX4,诱导脂质过氧化和细胞铁死亡,其抑制铁死亡效率比GPX4抑制剂的效率高5倍。将dGPX4封装在一种新型ROS可降解脂质纳米颗粒中合成dGPX4@401-TK-12,发现其能在癌细胞中选择性降解,从而释放dGPX4,增强癌细胞中GPX4的降解和铁死亡,有效抑制肿瘤生长而无明显不良反应。
除了癌症以外,泛素化对铁死亡的调控同样在其他疾病的治疗中发挥作用。肝脏缺血/再灌注以后可引起肝损伤和严重的细胞死亡,包括铁死亡,HUWE1可通过泛素化修饰TFR,使其发生蛋白酶体降解,从而调节铁代谢[14]。研究表明铁死亡参与到了帕金森综合征中,He等[27]总结了VDAC在帕金森综合征中的作用,认为联合使用药物抑制VDAC1的低聚化并促进VDAC1的泛素化可能是一种有效抑制帕金森综合征的方法,但目前VDAC2/3在帕金森综合征中的作用还有待进一步研究。
5 结语
泛素化是最常见的翻译后修饰之一,泛素化酶是一个庞大的家族,它通过蛋白酶体促进蛋白质降解,这一过程可以被去泛素化酶逆转。铁死亡已成为当前癌症治疗领域的热点,对其他疾病的治疗研究也越来越深入,泛素化和铁死亡对各种生理病理过程的重要性不容忽视,进一步探究两者的相互作用,对疾病治疗及其机制探究具有重大意义。
尽管铁死亡途径相关分子蛋白的泛素化修饰在调节铁死亡中发挥重要作用,但涉及的诸多机制还有待阐明,如:BAP1对SLC7A11表达的调节在H2A非泛素化突变体表达细胞中是否有变化?GPX4泛素化调节机制如何?VDAC1在PD患者和体外模型中表达水平为何不同?参与铁死亡发生发展的分子众多,随着研究的深入,关键的分子会逐渐明朗。目前通过靶向调节铁死亡相关分子泛素化水平治疗疾病的报道较少,且其中大部分针对铁死亡中的泛素化修饰的研究都聚焦于动物及细胞中的癌症治疗,真正用于临床疾病的治疗还有很长的路要走。
猜你喜欢
广州大学学报(自然科学版)(2019年1期)2019-05-07
华东师范大学学报(自然科学版)(2018年2期)2018-05-14
科学中国人(2017年36期)2017-06-09
天津科技大学学报(2016年1期)2016-02-28
湖北师范大学学报(自然科学版)(2015年2期)2016-01-10
中国病理生理杂志(2015年8期)2015-12-21
浙江大学学报(农业与生命科学版)(2015年4期)2015-12-15
中国医学科学院学报(2015年5期)2015-03-01
现代检验医学杂志(2015年2期)2015-02-06
四川生理科学杂志(2014年3期)2014-02-28
