
幽门螺杆菌诱导上皮-间质转化在胃癌中的作用机制
2024-02-01 09:38孙振灿周举坤许云鹏王玉平
协和医学杂志 2024年1期
孙振灿,周举坤,许云鹏,王 军,郑 亚,王玉平,姬 瑞,4
1兰州大学第一临床医学院,兰州 730000 兰州大学第一医院 2消化科 3消化内镜中心 4甘肃省消化系疾病临床医学研究中心,兰州 730000
胃癌是最常见的恶性肿瘤之一,居全球癌症发病排行第5位和死因排行第4位[1]。幽门螺杆菌 (Helicobacterpylori,Hp)是胃癌最重要的危险因素之一,一项荟萃分析显示,中国人群Hp感染率为49.6%[2]。Hp感染宿主后,激活宿主细胞中多种信号通路和蛋白,诱导胃上皮细胞发生上皮-间质转化 (epithelial-mesenchymal transition,EMT),而EMT异常调控则可导致胃癌发生[3-5],并增强肿瘤细胞迁移、侵袭,并参与肿瘤转移、化疗耐药等过程[6]。本文就Hp诱导胃上皮细胞发生EMT并导致胃癌的相关机制展开综述,以期为胃癌的早期诊断和靶向治疗提供新思路。
1 上皮细胞与间质细胞特征
人类上皮细胞和间质细胞均有特殊标志物[7],详见表1。EMT本身是一种正常的生理现象,参与胚胎发育和成人伤口愈合等过程。在EMT过程中,许多上皮细胞标志物表达降低,如ZO-1蛋白和E-钙黏蛋白,并诱导间质细胞标志物表达,如波形蛋白、N-钙黏蛋白、Snail蛋白和TWIST蛋白等。而当EMT异常调控时,上皮细胞极性丧失、形状变细长,细胞运动和侵袭能力增强。
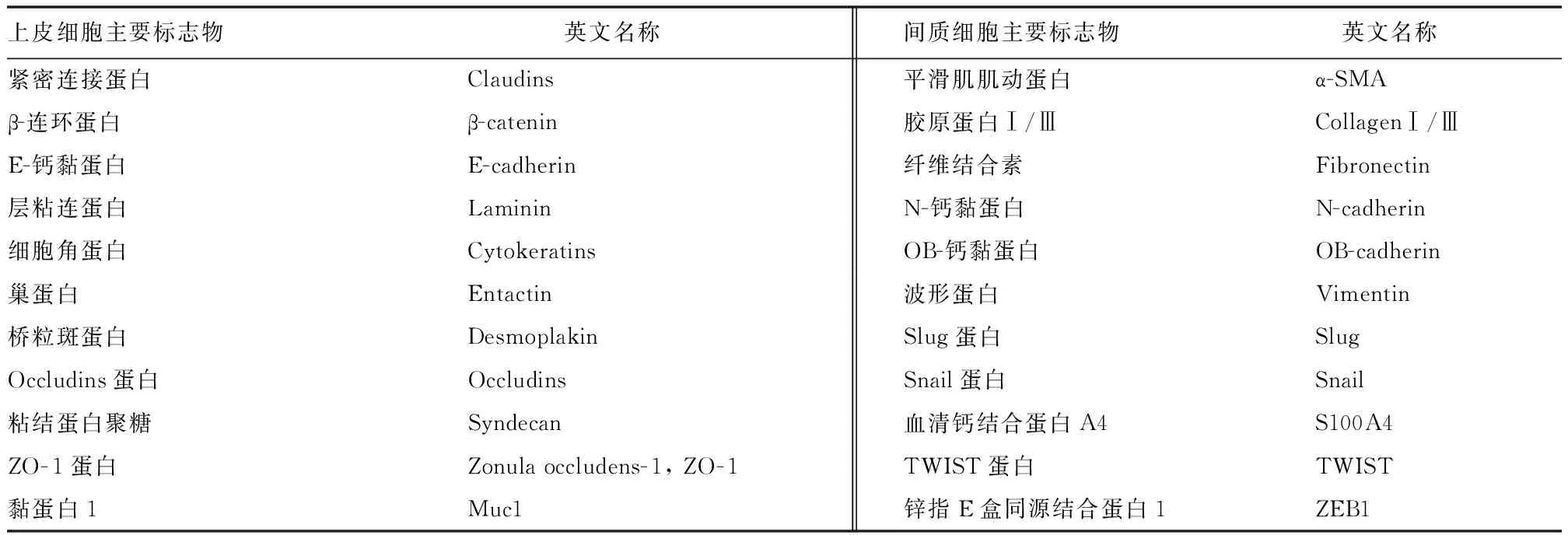
表1 上皮细胞及间质细胞主要标志物Tab.1 Main markers of epithelial and mesenchymal cells
2 Hp诱导EMT致胃癌相关机制
2.1 CagA阳性Hp诱导EMT相关机制
细胞毒素相关基因A(cytotoxin-associated gene A,CagA)是Hp最重要的毒力因子之一。CagA蛋白分子量为128~145 kDa,其羧基端有一段谷氨酸-脯氨酸-异亮氨酸-酪氨酸-丙氨酸 (Glu-Pro-Ile-Tyr-Ala,EPIYA)的氨基酸序列,决定CagA生物活性。Hp Cag致病岛 (cag pathogenicity island,CagPAI)编码CagA蛋白和一种大型膜相关转运蛋白复合体—Ⅳ型分泌系统 (Type Ⅳ secretion systems,T4SSs)。CagA蛋白可通过Ⅳ型分泌系统进入胃上皮细胞中。在萎缩性胃炎和胃癌中,CagA阳性Hp的致病性较CagA阴性Hp更强。
2.1.1 CagA诱导EMT
在胃上皮细胞内,CagA通过下调部分因子或蛋白诱导EMT,如糖原合成酶激酶-3 (glycogen synthase kinase-3,GSK-3)、蛋白激酶CK2β和程序性细胞死亡蛋白4 (programmed cell death factor 4,PDCD4)。CagA与GSK-3结合,使其转变为不溶性,从而降低GSK-3活性,诱导Snail介导的EMT[8]。Lee等[9]研究表明,CagA阳性菌株 (60190)相较于CagA阴性菌株感染的人胃腺癌上皮细胞(human gastric adenocar-cinoma epithelial,AGS)中E-钙黏蛋白和ZO-1减少,N-钙黏蛋白和Snail增加,且CagA诱导HDM2蛋白磷酸化导致CK2β降解,从而诱导EMT,促进胃癌发生发展。故CK2β可能是Hp感染导致胃癌EMT的关键介质,可作为胃癌治疗的分子靶点。胃上皮细胞内,Hp通过CagA调控PDCD4,进一步诱导胃癌细胞表达TWIST1,进而诱导EMT,促进胃癌发生发展[10]。
CagA也可通过激活部分蛋白或基因诱导EMT,如尾型同源盒基因1(caudal type homeobox 1,CDX1)、Yes相关蛋白 (yes-associated-protein,YAP)和水通道蛋白-5 (aquaporin 5,AQP5)。CDX1主要参与CagA促进胃慢性炎症向肠上皮化生的转化过程。Choi等[11]研究表明,CagA阳性Hp感染HFE145细胞后诱导CDX1过表达。张珊珊等[12]研究发现CDX1过表达的细胞中,E-钙黏蛋白表达减少,N-钙黏蛋白、波形蛋白表达增加,细胞侵袭及迁移能力均增强,表明Hp通过CagA调控CDX1诱导EMT。Choi等[11]利用二甲双胍处理CDX1过表达的HFE145细胞,并观察到CDX1表达降低,E-钙黏蛋白表达上调恢复,抑制了细胞侵袭和迁移,提示二甲双胍可逆转或抑制与CDX1相关的EMT。而CDX1可否作为胃癌靶向治疗的理想候选分子仍有待进一步研究,或许可使用抑制CDX1表达的药物逆转或抑制EMT,以防止肠上皮化生向胃癌的进一步发展。YAP是Hippo信号通路的一个元件,在细胞、肿瘤增殖方面发挥重要作用[13],Li等[14]研究表明,YAP在人胃癌组织中表达上调,且与肿瘤大小和转移相关。在CagA阳性Hp感染的胃上皮细胞内,YAP表达上调抑制E-钙黏蛋白表达,从而促进EMT,且E-钙黏蛋白抑制程度与慢性非萎缩性胃炎向胃癌的进展呈正相关,也进一步说明Hp感染诱导EMT对胃癌的发生发展具有重要影响。因此,YAP作为Hp诱导胃癌发生的一种新的分子靶点,或可为延缓胃癌发展提供新思路。Li等[15]的另一项研究表明,AQP5在人胃组织中的表达也随着慢性非萎缩性胃炎向胃癌的进展而逐渐增加,且CagA阳性Hp可通过激活MEK/ERK信号通路促进AQP5表达,从而诱导EMT发生,促进胃癌发生发展。
2.1.2 CagA诱导癌症干细胞特性
癌症干细胞是具有分化能力和自我更新能力的一种高致瘤性细胞,可以促进EMT和肿瘤生长,一直被认为是肿瘤扩散和复发的根源。Bessède等[16]研究发现,CagA阳性Hp感染胃上皮细胞后,干细胞标志物CD44显著增加,细胞表现出癌症干细胞特性,如 “蜂鸟表型”。在此过程中,间充质标志物Snail1蛋白、波形蛋白和ZEB1增加,诱导EMT发生,增加细胞迁移和侵袭能力。Choi等[11]研究进一步表明Hp CagA诱导癌症干细胞样表型的机制可能为胃上皮细胞中CDX1过表达,诱导CD44及性别决定区Y框蛋白2 (sex determining region Y-box 2,SOX2)、八聚体结合转录因子4(octamer-binding transcription factor 4,OCT4)和c-Myc蛋白等癌症干细胞特性相关蛋白表达增加。在慢性CagA阳性Hp感染过程中,化学致癌物甲基硝基亚硝基胍可协同Hp增加OCT4、c-Myc、SOX2表达[17],导致胃黏膜上皮细胞获得癌症干细胞样特性,诱导EMT,增强胃癌细胞侵袭和迁移。
2.2 Hp与肿瘤坏死因子α诱导蛋白
肿瘤坏死因子α(tumor necrosis factor-α,TNF-α)是一种内源性肿瘤启动子,与Hp诱导胃内炎症相关。有研究首次发现Hp产生TNF-α诱导蛋白(tumor necrosis factor-α-inducing protein,Tipα)使MKN-1细胞表达波形蛋白,形成丝状伪足和侵袭性表型,促进胃癌细胞伸长并向细胞外迁移[18],这表明Hp Tipα蛋白诱导EMT,促进胃部炎症向胃癌转化。Chen等[19]研究表明,Tipα可通过白细胞介素(interleukin,IL)-6/STAT3通路导致SGC7901细胞中E-钙黏蛋白下调,波形蛋白和N-钙黏蛋白上调,诱导EMT,促进胃癌细胞增殖和迁移。近年来研究显示Tipα通过激活Wnt/β-catenin通路,诱导胃癌细胞发生EMT[20- 21]。Tipα作为一种新发现的可促进Hp炎症和致癌作用的蛋白,未来或可作为胃癌治疗的新靶点。
2.3 Hp与间充质干细胞
间充质干细胞(mesenchymal stem cells,MSCs)是一种可分化为多种细胞并维持自我更新能力的细胞。胃癌MSCs通过大量分泌IL-8和IL-6促进胃癌微环境中M2型巨噬细胞极化,增加纤维结合素、波形蛋白和Snail2蛋白表达,降低E-钙黏蛋白表达,诱导EMT发生[22]。Zhang等[23]研究提示Hp感染MSCs后,分泌IL-6、IL-8和粒细胞集落刺激因子 (granulocyte-macrophage colony-stimulating factor,GM-CSF)等细胞因子,且E-钙黏蛋白表达下降,N-钙黏蛋白和波形蛋白表达上调。提示Hp可通过MSCs诱导EMT发生,从而促进胃癌细胞迁移。
2.4 Hp与癌症相关成纤维细胞
Hp感染会导致成纤维细胞和肌成纤维细胞分化为癌症相关成纤维细胞(cancer-associated fibroblasts,CAFs)[24-25],诱导EMT发生,促进细胞癌变。CAFs释放致癌因子和促炎因子对胃癌的发生、发展和转移至关重要。Hp感染成纤维细胞会导致成纤维细胞激活蛋白(fibroblast activation protein,FAP)、成纤维细胞表面蛋白(fibroblast specific protein,FSP)和转化生长因子-β(transforming growth factor-β,TGF-β)过表达,并增加促炎因子IL-6、IL-8、环氧合酶-2(cyclooxygenase-2,COX-2)和血清基质衍生因子1(stromal-derived factor 1,SDF-1)表达,诱导成纤维细胞形态改变,细胞中α-SMA、胶原蛋白Ⅰ、胶原蛋白Ⅲ、波形蛋白和N-钙黏蛋白表达显著增加[26-27]。此类成纤维细胞会导致正常胃上皮细胞形态变细长,细胞中TWIST、Snail、β1-整合素和COX-2表达增加、E-钙黏蛋白表达下调[26],提示EMT的发生。
2.5 miRNA
miRNA是一类非编码RNA,通过与mRNA 3’非翻译区结合,导致mRNA降解或抑制其翻译,从而调控基因表达。在胃癌发生发展过程中,Hp可诱导部分miRNA过表达,促进EMT发生。Zhu等[28]研究发现,Hp可导致胃上皮细胞miR-584和miR-1290过表达,从而下调靶基因Foxa1,导致E-钙黏蛋白表达降低,诱导EMT。后续研究中,研究者逐渐发现Hp可诱导miR-29a-3p、miR-543和miR-155等多种miRNA过表达,在此过程中,胃上皮细胞中E-钙黏蛋白减少,Snail蛋白、波形蛋白和N-钙黏蛋白增多,诱导EMT[29-31]。这说明miRNA有调节EMT的作用,具有促进胃癌细胞迁移和侵袭能力。
Hp除通过诱导上述miRNA过表达进一步诱导EMT,有研究发现部分miRNA过表达可起到逆转或抑制EMT的作用。已有研究证明miR-490-3p减少是Hp相关性胃癌的独立危险因素[32]。Shen等[33]研究表明,miR-490-3p过表达可抑制EMT。Huang等[34]研究发现,miR-134过表达可能通过靶向FoxM1抑制SGC-7901细胞增殖、侵袭和EMT。上述研究提示,miRNA可抑制EMT,降低胃癌细胞的侵袭和迁移能力,延缓胃癌进程。因此,miRNA或许是治疗Hp相关胃癌的关键靶点。
2.6 其他因子
除上述蛋白或基因与Hp诱导EMT密切相关,还有其他转录因子、蛋白也参与Hp诱导EMT的过程,但此类研究相对较少,如外泌体、GSK-3β、Afadin蛋白、青霉素结合蛋白1A(penicillin-binding protein1A,PBP1A)、溶酶体相关4次跨膜蛋白B(lysosomal-associated protein transmembrane 4b,LAPTM4B)和核转录因子-E2相关因子2(nuclear factor-like 2,Nrf 2)等。
胃癌细胞将含有活化间充质上皮转移因子的外泌体递送至巨噬细胞,进一步刺激巨噬细胞促进肿瘤生长。在此过程中可观察到胃癌细胞中E-钙黏蛋白表达水平下降,波形蛋白和Snail蛋白表达显著增加,表明Hp诱导的外泌体可能参与诱导EMT的过程[35]。Ouyang等[36]研究发现,Hp通过AKT/GSK-3β信号通路调控GES-1细胞发生EMT。Marques等[37]观察到Hp下调MKN74和NCI-N87细胞系中Afadin蛋白表达,诱导EMT。Huang等[34]研究显示,PBP1A会导致SGC7901细胞中E-钙黏蛋白减少,平滑肌肌动蛋白增多。Zhou等[38]研究显示,Hp诱导LAPTM4B上调,导致细胞中E-钙黏蛋白和ZO-1蛋白表达下调,N-钙黏蛋白和波形蛋白表达上调,诱导EMT。Hp还会降低Nrf2活性,导致上皮细胞标志物减少、间质细胞标志物ZEB1和Snail蛋白增加[39],促进EMT发生。以上研究均表明Hp可通过多种方式诱导EMT发生,最终导致胃癌细胞增殖、迁移和侵袭。另有研究表明,在Hp感染早期,使用天然Nrf 2激活物可减少Hp相关胃炎和氧化应激,延缓炎症向胃癌发展[40-41]。
3 小结与展望
Hp在诱导胃上皮细胞发生EMT并导致胃癌发生发展过程中发挥重要作用。Hp分泌的毒力因子、细胞成分通过不同机制诱导EMT发生,进而影响胃炎向胃癌进展的速度和最终结局。虽然Hp在20年前就已经被确定为胃癌相关的I类致癌物,但其导致胃癌发生的机制仍是目前研究的热点,虽然大多数研究已阐明Hp诱导EMT发生的机制,但仍有部分研究仅在实验中观察到Hp导致胃上皮细胞发生EMT,具体机制仍需进一步探索,以期发现可在Hp致胃癌过程中给予早期干预的新方法,减少胃癌发生发展,同时也为胃癌靶向治疗提供新思路。
作者贡献:孙振灿、周举坤、许云鹏负责文献检索和论文撰写;王军、郑亚、王玉平、姬瑞负责论文修订。
利益冲突:所有作者均声明不存在利益冲突
猜你喜欢
今日农业(2022年13期)2022-09-15
昆明医科大学学报(2021年8期)2021-08-13
云南医药(2021年3期)2021-07-21
生物学通报(2020年10期)2020-08-13
知识经济·中国直销(2017年10期)2017-11-07
中华老年多器官疾病杂志(2016年9期)2016-04-28
中国现代医学杂志(2015年26期)2015-12-23
医学研究杂志(2015年7期)2015-06-22
中国当代医药(2015年33期)2015-03-01
现代检验医学杂志(2015年6期)2015-02-06
