
剪切增稠液微胶囊的制备与性能研究
2024-01-25 10:40赵炳乾倪叶舟俞科静陈坤林
中国塑料 2024年1期
俞 波,赵炳乾,倪叶舟,钱 坤,俞科静,陈坤林
(江南大学生态纺织教育部重点实验室,江苏 无锡 214122)
0 前言
高性能防护材料在体育竞技[1]、军事战争[2]和复杂环境[3]等领域中扮演着重要角色。在早期阶段,防护材料主要采用金属和陶瓷等刚性材料来抵御外部冲击,但它们相对笨重且缺乏柔韧性和舒适性,限制了其广泛应用。
STF 作为一种质轻且抗冲击性能好的新型智能材料,一旦剪切速率超过其临界剪切速率,黏度可以增加数个数量级,并在去除剪切应力时恢复到初始状态[4]。STF 因其对外部冲击载荷的可逆敏感响应而备受关注。目前剪切增稠材料通常分为剪切增稠凝胶(STG)和STF,STG 和STF 作为抗冲击材料都显示出巨大的前景[5]。STG 是一种低交联度聚硼硅氧烷,而STF 是一种由高含量分散相颗粒和液体分散介质组成的非牛顿悬浮液,与液体相比承受更高的外力,表现出与固体不同的可变形状特征,因此STF在减振、人体防护和表面抛光等领域具有广泛的应用前景。
然而STF 本质上是一种流体,对环境敏感。现阶段STF 分散介质多采用聚乙二醇(PEG)等吸湿性较强的溶剂,在实际使用过程中会因为吸收空气中的水分而使分散体系剪切增稠性能降低。此外,由于STF的高黏度、多组分,与织物复合过程中存在脱落以及难以处理或集成到结构中等问题[6],因此在实际应用中需要对其适当封装。Zhang 等[6]利用由分子量为600 的5 %聚乙烯亚胺(PEI)和10 %乙二醇(EDO)和85 %STF 组成的溶液通过针管注射器注射到由二异氰酸酯预聚物Suprasee 2644、甲苯和氯仿组成的反应溶液中,通过该方法制得的胶囊平均直径为(2.7±0.2)mm。Liu 等[7]利用孔口凝固浴法包封STF,研究不同氯化钙浓度、海藻酸钠浓度和表面活性剂用量,以胶囊强度为指标,得出最优制备工艺参数,通过该方法得到的胶囊平均直径在1.93 mm,胶囊的热稳定性也有所改善。然而这些文献中制备的胶囊尺寸都在宏观的毫米级别,尺寸过大,使得STF胶囊难以分散到基质材料中[8]。
基于此,本文提出了一种单凝聚法壳聚糖包覆微米级STF MCs 的方法,可控制微胶囊的直径在几微米范围内。首先在Span 80 和Tween 80 的乳化作用下,STF 被充分分散在正己烷中,形成初始乳液;然后将初始乳液与曲拉通X-100 溶液充分分散,形成复乳液,最后将壳聚糖溶液加入上述复乳液中,形成STF微胶囊。壳聚糖作为壁材具备优异的成膜性能,可以尽可能的保持STF的流变性能,能有效提高材料的抗冲击性能。
1 实验部分
1.1 主要原料
二氧化硅(SiO2),650 nm,国药集团化学试剂有限公司;
碳纳米管(CNTs),纯度>97 %,深圳市纳米科技港口有限公司;
PEG,化学纯,国药集团化学试剂有限公司;
正己烷,分析纯,纯度≥97 %,国药集团化学试剂有限公司;
Span 80,纯度≥化学纯(沪试),国药集团化学试剂有限公司;
Tween 80,分析纯,纯度≥分析纯,上海泰坦科技股份有限公司;
曲拉通X-100,纯度>98 %,上海麦克林生化科技有限公司;
壳聚糖,分析纯,纯度≥中黏黏度,200~400 mPa·s,北京伊诺凯科技有限公司;
乙酸,分析纯(沪试),纯度≥99.5 %,国药集团化学试剂有限公司。
1.2 主要设备及仪器
光学显微镜(PLM),Motic,麦克奥迪实业集团有限公司;
扫描电子显微镜(SEM),S4800,日本株式会社日立高新技术;
X 射线能谱仪(EDS),ELECT PLUS,美国EDAX公司;
激光粒度分析仪,Bettersize2600,丹东百特仪器有限公司;
旋转流变仪,Physica MCR301,安东帕(上海)商贸有限公司;
热失重分析仪(TG),Q500,美国TA 仪器有限公司;
傅里叶变换红外光谱仪(FTIR),NICOLET.is10,美国赛默飞世尔科技(中国)有限公司。
1.3 样品制备
SiO2基STF 及掺杂CNTs STF 的制备:为了平衡剪切增稠性能和液体流动性间的关系[9],本文选用了粒径较小的SiO2粒子作为体系的分散相粒子,分子量相对适中的PEG作为体系的分散介质。为了进一步增强STF 的剪切增稠性能,还选用了与PEG 具有相似的长链状结构的碳纳米管作为填料引入SiO2基STF 构多相STF。首先将SiO2粒子放入100 ℃烘箱中进行24 h的烘干处理,以确保粒子在制备待用前处于干燥状态,随后称取一定质量比的SiO2粒子、PEG及无水乙醇,将以上物料通过高速剪切乳化机进行2 h 高速剪切处理和超声处理20 min,随后将制得的SiO2基STF 在60 ℃的真空烘箱干燥处理12 h,以去除无水乙醇及气泡。掺杂CNTs STF 的制备过程与SiO2基STF 基本一致,区别在于物料中引入一定量的CNTs,增加剪切乳化时间至3 h 以及增加超声处理时间至30 min,以确保CNTs可以作为多相分散相充分分散在PEG中。
SiO2基STF MCs 及掺杂CNTs STF MCs 的制备:在一定量上述制得的SiO2基STF 中加入适量去离子水,待SiO2基STF 溶解形成水溶液,再将一定量正己烷作为油相与上述水相混合,在此基础上加入适量的乳化剂,并进行超声乳化30 min 形成初乳液,再将初乳液分散在一定量去离子水中,加入适量曲拉通X-100,超声乳化30 min,获得复乳液。然后将乳化完全的上述复乳液加入到壳聚糖酸性溶液中,超声乳化10 min,在30 ℃下保温反应1 h,最后升温至一定温度保温反应6 h,制得所需SiO2基STF MCs。掺杂CNTs STF MCs 的制备方式与SiO2基STF MCs 的制备方法基本一致,区别在于将芯材SiO2基STF 替换成掺杂CNTs STF,在此不多赘述。
1.4 性能测试与结构表征
流变性能测试:采用平行板式旋转流变仪,在25 ℃下对STF体系进行流变性能测试,其中,选用平板夹具的直径为50 mm(型号为PP50),随即启动仪器配套软件开始测试,软件中会随之出现剪切速率、黏度、模量等数据;
微观形貌分析及EDS 分析:使用PLM 和SEM 观察样品表面微观形貌,并拍摄微胶囊的显微镜照片;利用ESD 测试微胶囊表面的元素组成,测试电压为10 kV;
FTIR 分析:根据KBr 压片法,使用FTIR 对SiO2基STF MCs 及掺杂CNTs STF MCs、微胶囊壳层和芯材SiO2基STF 及掺杂CNTs STF 进行FTIR 分析,扫描范围为4 000~500 cm-1,分辨率为0.5 cm-1;
微胶囊包埋率分析:微胶囊的包埋率,又称包封率,是指制得的微胶囊产物中芯材的实际质量与芯材理论质量的百分比。微胶囊的包埋率越高,代表制备过程中生产效率越高,从而经济效益越好。微胶囊包埋率的测定方法有很多,常用的方法包含溶剂洗涤法、TG 曲线分析法和差示扫描量热量仪器(DSC)分析法。DSC 分析法是用DSC 测得微胶囊芯材和微胶囊的相变潜热来测得微胶囊的包埋率。而TG 法则是根据TG 曲线来获得微胶囊包埋率大小,将制备得到的样品称取5 mg 左右放入坩埚中,在氮气气氛下,从30 ℃以20 ℃/min 的升温速率增加至800 ℃。根据TG 曲线和式(1)测得微胶囊的包埋率:
式中E——微胶囊包埋率,%
m——微胶囊产物质量,mg
W——芯材占样品总质量,mg,由TG曲线可得
M——理论所得芯材质量,mg
2 结果与讨论
2.1 STF MCs工艺参数研究
在单凝聚法制备微胶囊的合成过程中,对复合材料性能产生影响的因素有很多,本文重点研究了STF HLB 值、乳化剂含量、核壳比、搅拌速度、反应温度、STF MCs 在分散液中的浓度对微胶囊微观形貌和粒径分布的影响
2.1.1 STF HLB值测定
乳化剂的使用可以减少油水两相的界面张力,促进乳液中胶束的形成。由于不同乳化剂的HLB 值(指表面活性剂分子中亲水和亲油基团对油或水的综合亲和力)的差异,对最终所形成的乳液的液滴尺寸和形态均会产生影响,从而最终影响乳液的稳定性及后续微胶囊的性能。当所需乳化剂HLB 值和芯材所需的HLB 值接近时,二者混合后可得到相对稳定的乳液。此外,复合乳化剂的乳化效果一般优于单种乳化剂,结构相似的乳化剂混合使用时,其协同效应比较明显,因此本实验除了选取单个乳化剂,还选择了Span 80 和Tween 80 复配的乳化剂,基于此,得到了不同HLB 值的乳化剂并制备得到了W/O 初乳液。不同种乳化剂制备的初乳液性能评价如表1所示。
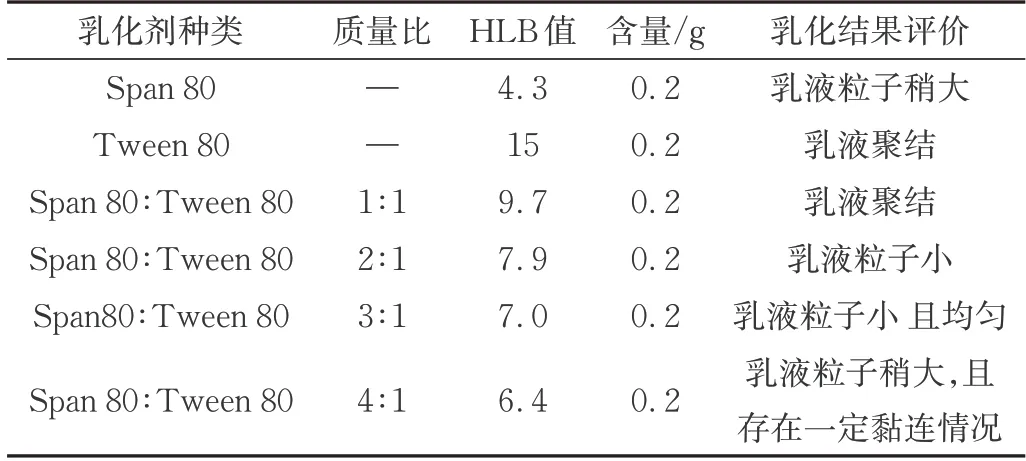
表1 不同种乳化剂制备的初乳液性能评价Tab.1 Evaluation of the properties of colostrums prepared from different emulsifiers
由图1 可知,当乳化剂分别采用Tween80 和Span80 与Tween80 在1∶1 复配的情况下,乳液在超声时会聚结,不能形成稳定的乳液。这可能与所选用乳化剂的HLB 值不适合W/O 体系有关。当乳化剂选用Span 80 与Tween 80 复配且复配比分别为2∶1 和3∶1所形成的W/O 乳液体系具有相似的粒子尺寸和大小,而当分别选用单一乳化剂Span80 以及Span 80 与Tween 80 复配乳化剂且复配比为3:1 时,粒子形态较好且尺寸更小。因此,最终选择乳化剂为Span 80 与Tween 80 复配,质量为3∶1 时,进行W/O 乳液的制备。

图1 不同种乳化剂制备的W/O初乳的PLM照片Fig.1 Polarizing microscopy of W/O colostrum prepared from different emulsifiers
2.1.2 乳化剂含量
除了乳化剂种类外,其含量对微胶囊的制备极其重要[10]。保持其他条件相同的情况下,改变乳化剂的含量,制得不同乳化剂含量下微胶囊初乳液的粒径随含量的变化如图2所示。可以看出,随着Span 80 与Tween 80 复配乳化剂含量的增加,初乳液的粒径整体呈现减小的趋势,而随着乳化剂的含量从5 %增加至13 %,初乳液的粒径则由1 354 nm 减小至345 nm,这是因为当乳化剂含量不足时,粒子间的斥力不足以克服液滴间的引力时,而发生团聚现象,最终不能形成稳定的乳液,致使初乳液的粒径较大,然而随着乳化剂的含量增加至11 %时,粒子分散更充分,制得的乳液粒径可减小至447 nm,当乳化剂进一步增加至13 %时,粒径基本不变,因此制备初乳液的乳化剂合适含量为11 %。
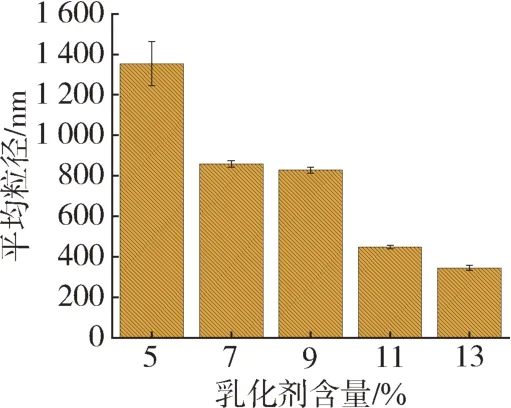
图2 乳化剂含量对微胶囊初乳液粒径的影响Fig.2 Effect of emulsifier content on particle size of microencapsulated colostrum
2.1.3 核壳比
在微胶囊合成过程中,壳材与芯材的质量比也会影响微胶囊的粒径大小、芯材的包埋率和微胶囊产率[11]。为探讨核壳比对微胶囊粒径和分布,保持其他条件相同的情况下,改变核壳比,制得不同核壳比下微胶囊的粒径随核壳比的变化如图3所示。考虑到包埋率和微胶囊产率的影响,本文设计了系列核壳比,分别为1∶1、1∶2和1∶3。由图3可知,随着核壳比的减小(由1∶1 减小至1∶3),即壳材含量的增加,微胶囊平均粒径由2 950 nm 增加至3 210 nm。壳材质量增加,包埋率亦随之增加。壳材变厚,粒径也随之增大。此外,考虑到工业生产的需求,还设计了核壳比为3∶1 和2∶1 的核壳比方案,由图可知,随着核壳比由3∶1 降至2∶1,微胶囊平均粒径由1 600 nm 增加至1 800 nm,增大趋势较为明显,这说明当壳材比为2∶1 时,微胶囊包裹更加充分。综合包埋率和工业生产的需求,制备微胶囊合适的壳材比选为2∶1。
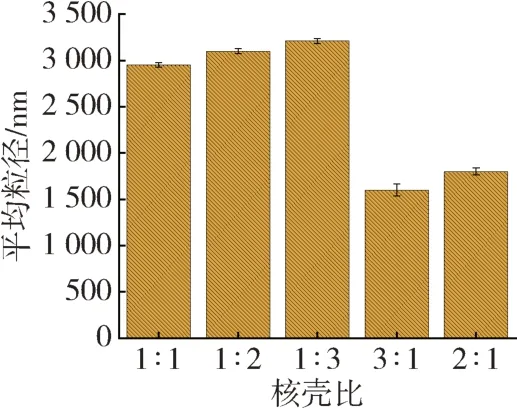
图3 核壳比对微胶囊平均粒径的影响Fig.3 Effect of core-shell ratio on the average particle size of microcapsules
2.1.4 搅拌速度
搅拌速度亦是微胶囊合成过程中对其粒径、粒径分布以及壁材沉积形成控制的重要影响因素之一。若搅拌速度过低,会导致乳液无法混合均匀,产生黏结甚至团聚等现象,对后续微胶囊的形成产生较大的影响,若搅拌速度过高,则有可能导致乳液喷溅从而影响微胶囊的合成。搅拌速度对微胶囊的粒径及其分布的影响如表2所示。可以看出,随着搅拌速度的增加,微胶囊的平均粒径及聚合物分散指数(PDI)都呈现出减小的趋势,这是因为随着搅拌速度的增加,其剪切应力也会随之增大,而当粒子表面张力不足以抵消搅拌过程中所产生的剪切应力时,微胶囊粒子就会被分散得更加充分。当搅拌速度达到1 000 r/min 时,乳液的平均粒径减小趋势明显下降,这可能是由于高搅拌速度过高导致了更高的剪切速率所引起。这可使得部分反应乳液被离心力甩到容器壁处,致使粒子间的斥力不足以克服相互之间的引力。因此,微胶囊合成过程中搅拌速度需控制在600 r/min,这对微胶囊粒径及粒径分布的控制较为合适。

表2 搅拌速度对微胶囊粒径及粒径分布的影响Tab.2 Effect of stirring speed on particle size and particle size distribution of the microcapsules
2.1.5 反应温度
反应温度对相分离法制备壳聚糖微胶囊的影响也较大,当反应温度较低时,对反应体系pH 的影响不大,而壳聚糖从溶液中析出的速度则较慢,反应亦不完全,从而生成的产物较少;但是当温度过高时,壳聚糖中的氨基基团易受到氧化的影响。因而,应在适当的反应温度下,所形成的微胶囊性能较好且在形成微胶囊的过程中速度也较快。因此本实验的反应温度控制在60 ℃。
2.1.6 STF MCs在分散液中的浓度
考虑到后续工作的需要,本文讨论了STF MCs在微胶囊分散液体系中的浓度,即通过调节体系O/W比(1∶1、1∶2、1∶3、1∶4)调控体系的浓度。制得的系列不同油水比微胶囊的粒径如图4所示。可以看出,随着整个体系O/W 比的减小,微胶囊分散液的粒径也随之减小,然而此时STF MCs 在微胶囊分散液中的浓度亦随之减小。考虑到平衡后续实验室工作中制样的效果和工业生产需求,即微胶囊分散液中的STF MCs 的浓度和粒径大小,将后期的制备工艺中的O/W 比定为1∶2。
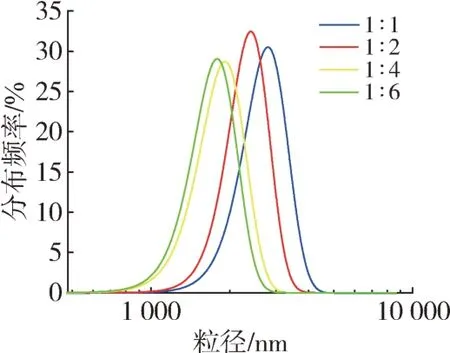
图4 O/W比对微胶囊粒径的影响Fig.4 Effect of oil-water ratio on particle size of the microcapsules
2.2 流变性能测试
为研究微胶囊中剪切增稠液的剪切增稠性能,除了选用制备STF 最常见的分散相二氧化硅(SiO2)基础体系以外,还进一步通过添加少量的碳纳米管(CNTs)来调控STF的流变性能[12],如图5所示,SiO2基STF即基础体系的初始黏度为4.6 Pa·s,临界剪切速率为2.6 s-1,峰值黏度为157.2 Pa·s,而掺杂CNTs STF 体系的初始黏度为9.2 Pa·s,临界剪切速率为1.36 s-1,峰值黏度为294.3 Pa·s。相较不掺杂CNTs 的STF 基础体系而言,掺杂CNTs的STF 体系初始黏度有所增加,临界剪切速率更小,黏度突变更快,峰值黏度增加近一倍。由此可见,CNTs的引入显著增强了体系的剪切增稠效应,进一步提高了在工程应用中的防护性能。这可能与CNTs 分子链结构有关:(1)CNTs 与PEG 具有相似的的分子长链结构,二者间的流体润滑力更强,流体润滑力破坏了颗粒原有的二维层状排列[13],从而使得固体颗粒堵塞并形成颗粒团簇。(2)CNTs 的网状结构有助于当剪切速率进一步增加到临界值,流体力明显高于粒子与布朗运动之间的分子间时,进一步促使系统中不稳定的官能团迅速聚集,从而系统的黏度进一步增加,出现剪切增稠现象。(3)在较高剪切速率下,CNTs的刚性使其能够承受更大的接触力,根据摩擦接触理论[14],当粒子之间的法向接触力较高时,粒子之间的流体膜被破坏,粒子之间的接触增加,粒子之间的接触力和摩擦力其主要作用。因此CNTs 具有更为显著的剪切增稠效应。
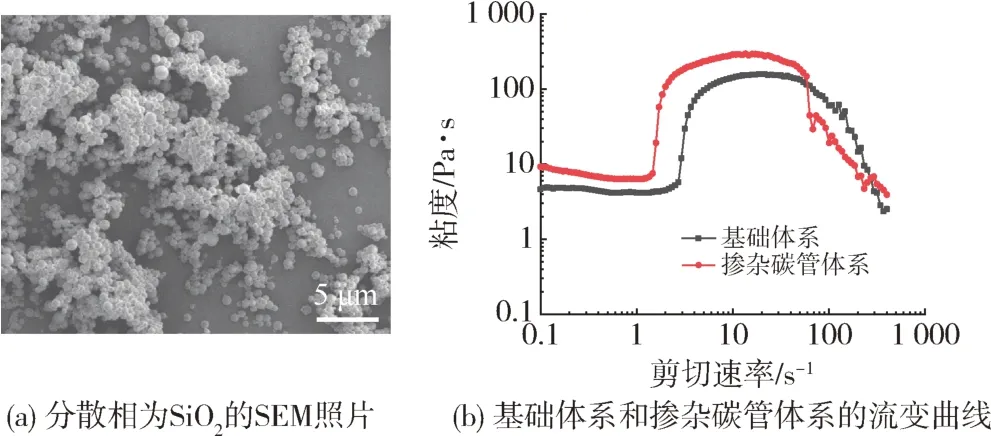
图5 分散相为SiO2的SEM照片以及基础体系和掺杂碳管体系流变曲线Fig.5 SEM of SiO2 and rheological diagrams of the base system and the doped carbon tube system
2.3 微观形貌及EDS分析
以壳聚糖为壳材,STF 作为芯材,按照上述确定的制备工艺,并采用相分离法制得了STF 微胶囊。分别通过PLM 和SEM 观察微胶囊的形貌,并利用Adobe Acrobat XI Pro 软件统计3 张SEM 照片中微胶囊的粒径,并取其平均分布。
从图6(a)的PLM 照片可以看到,制得的微胶囊为较规整的球形,且分散相对较为均一。进一步从6(b)、(c)和(d)中可以看出,微胶囊为规则的球形,粒径分布较为均匀且平均粒径为3 μm 左右,这些观测结果与光学显微镜测试的结果相一致。
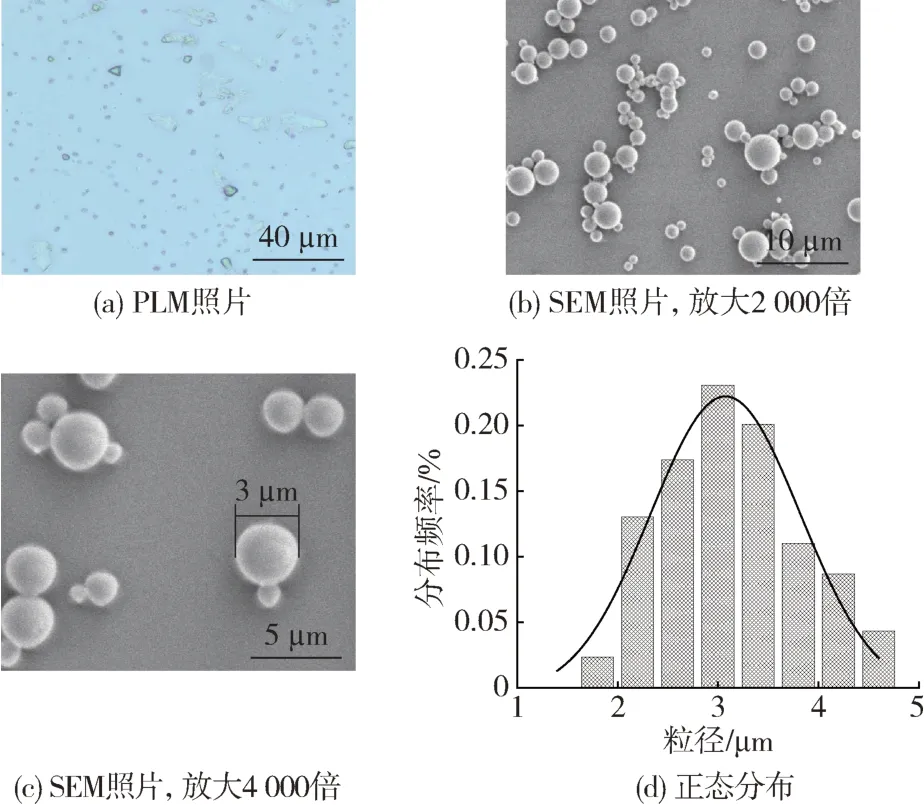
图6 微胶囊的微观形貌和正态分布图Fig.6 Microscopic morphology and normal distribution of the microcapsules
通过EDS表征对微胶囊表面的化学元素组成进行分析。从图7 可以看出,微胶囊表面主要由 C、N、O 3 种化学元素组成,其中N 元素含量约为11.5 %,而N元素的存在也说明壳聚糖是微胶囊的主要壳材[15]。
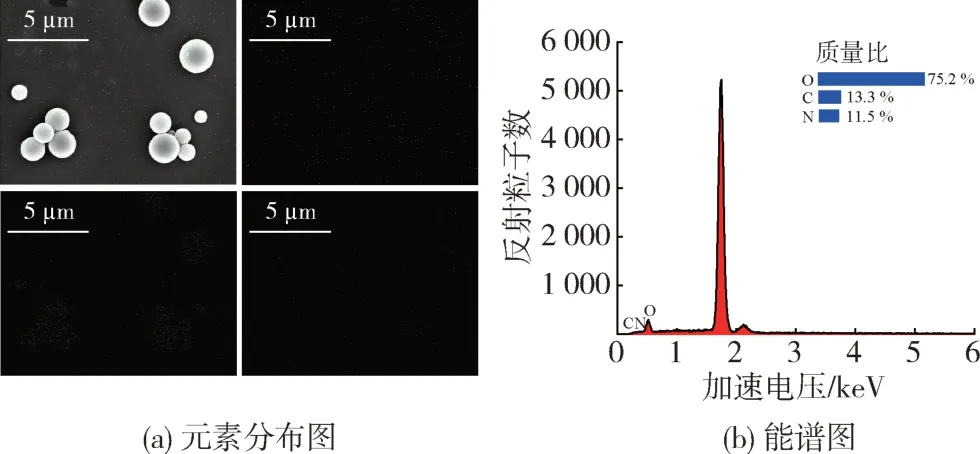
图7 微胶囊的EDS能谱分析Fig.7 EDS spectroscopy analysis of the microcapsules
2.4 FTIR分析
进一步对芯材、壁材和微胶囊进行FTIR测试。从图8可以看到,STF MCs、SiO2基STF、CS、掺杂CNTs基STF MCs、掺杂CNTs 基STF 在3 500 cm-1附近都存在一个明显的宽峰,其中STF MCs、CS、掺杂CNTs基STF MCs、掺杂CNTs 基STF 在该波长范围内主要是由于材料中—OH 伸缩振动引起的,SiO2基STF 中为—OH 和—NH 形成氢键的伸缩振动吸收峰;芯材的多个特征峰位置在微胶囊处均有体现。在STF MCs和SiO2基STF 曲线中,2980 cm-1处为C—H 的伸缩振动吸收峰,1 650 cm-1附近处为水的H—OH 弯曲振动峰,1 100 cm-1处的强峰为Si—O—Si 的反对称伸缩振动峰,814 cm-1处为Si—O 的对称伸缩振动峰,但在以上几个峰位置处,STF MCs 中SiO2基STF MCs 的拉伸明显减弱即峰的强度明显减弱,这可能是因为STF被壳聚糖包上后,限制了STF分子中原子的振动,因此峰值存在不同程度的偏移。芯材的FTIR 光谱大部分特征峰位置都在微胶囊处均有所体现,结合前面EDS表征,可以证明壳聚糖已成功吸附在芯材液滴表面。
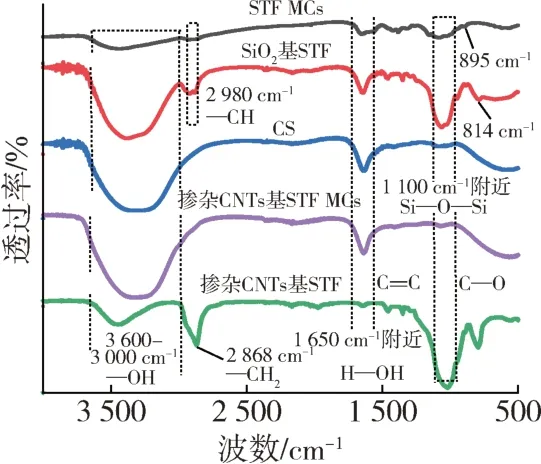
图8 微胶囊及其芯材、壁材的FTIR谱图Fig.8 FTIR of the microcapsules,their core materials and wall materials
2.5 TG分析
进一步利用TG 对微胶囊进行分析。如图9所示,样品的TG 曲线大致可分为3 个失重阶段,第一个失重阶段主要是微胶囊内残余水分挥发所致;而从250~400 ℃的第二失重阶段主要为壳聚糖壳材的分解所致;第三阶段温度从400 ℃升温后,样品质量不再发生变化,没有明显的失重现象,说明壳材壳聚糖和芯材都已经分解完成,其中掺杂CNTs微胶囊和不含CNTs微胶囊约占样品总质量的88 %和77 %,计算得知掺杂CNTs 微胶囊和不含CNTs 微胶囊的包埋率分别为59 %和62 %。
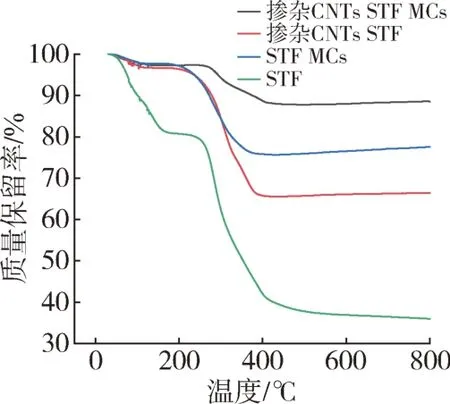
图9 样品的TG曲线Fig.9 TG curves of the sample
此外,与芯材无论是掺杂或未掺杂CNTs 的STF相比,它们的微胶囊的开始分解温度往往更高,这说明壁材对芯材起到了良好的保护作用,提升了芯材无论是掺杂或未掺杂CNTs的STF的热稳定性。
3 结论
(1)首先通过单因素实验研究确定了微胶囊的制备工艺参数:分散剂种类为Span 80 与Tween 80 复配且复配比为3∶1,乳化剂含量为11 %,核壳比为2∶1,搅拌速度为600 r/min,反应温度为60 ℃,体系的油水比为1∶2,在以上优化的制备工艺条件下,制得的STF MCs 具有较为规整的球形、粒径分布较为均匀且集中在3 μm左右;
(2)芯材和微胶囊乳液的FTIR 谱图大部分都一致,表明壳聚糖成功吸附在芯材液滴表面即STF 被成功包封上;壁材对芯材STF起到了保护作用,提升了芯材STF 的热稳定性;碳纳米管进一步的引入可改善STF 的流变性能,掺杂CNTs 的STF 体系临界剪切速率更小,黏度突变更快,峰值黏度增加近一倍;
(3)这些生产具有高抗冲击性的STF MCs的研究结果为设计和制造多功能抗冲击材料提供了一种新的设计思路。
猜你喜欢
天津科技(2022年7期)2022-07-29
当代水产(2022年2期)2022-04-26
天津科技(2021年7期)2021-07-29
原子与分子物理学报(2021年2期)2021-03-29
天津科技(2020年7期)2020-07-31
江西建材(2018年4期)2018-04-10
西安工程大学学报(2016年6期)2017-01-15
材料科学与工程学报(2016年1期)2017-01-15
湖北师范大学学报(自然科学版)(2015年1期)2016-01-10
中国造纸学报(2015年1期)2015-12-16
