
High glucose reduces Nrf2-dependent cRAGE release and enhances inf lammasomedependent IL-1β production in monocytes: the modulatory effects of EGCG
2024-01-24 01:11ChiHaoWuYinHsuanChangChinLinHsuShengYiChenGowChinYen
食品科学与人类健康(英文) 2024年3期
Chi-Hao Wu, Yin-Hsuan Chang, Chin-Lin Hsu, Sheng-Yi Chen, Gow-Chin Yen,
a Graduate Programs of Nutrition Science, School of Life Science, Taiwan Normal University, Taipei 106209, Taiwan, China
b Department of Food Science and Biotechnology, Chung Hsing University, Taichung 40227, Taiwan, China
c School of Nutrition, Chung Shan Medical University, Taichung 40201, Taiwan, China
Keywords: Epigallocatechin gallate (EGCG)Inf lammasome Nuclear factor erythroid 2-related factor 2 (Nrf2)Receptor for advanced glycation end products (RAGE)Soluble RAGE (sRAGE)
ABSTRACT Soluble receptor for advanced glycation end products (sRAGE) acts as a decoy sequestering of RAGE ligands, thus preventing the activation of the ligand-RAGE axis linking human diseases. However, the molecular mechanisms underlying sRAGE remain unclear. In this study, THP-1 monocytes were cultured in normal glucose (NG, 5.5 mmol/L) and high glucose (HG, 15 mmol/L) to investigate the effects of diabetesrelevant glucose concentrations on sRAGE and interleukin-1β (IL-1β) secretion. The modulatory effects of epigallocatechin gallate (EGCG) in response to HG challenge were also evaluated. HG enhanced intracellular reactive oxygen species (ROS) generation and RAGE expression. The secretion of sRAGE, including esRAGE and cRAGE, was reduced under HG conditions, together with the downregulation of a disintegrin and metallopeptidase 10 (ADAM10) and nuclear factor erythroid 2-related factor 2 (Nrf2) nuclear translocation.Mechanistically, the HG effects were counteracted by siRAGE and exacerbated by siNrf2. Chromatin immunoprecipitation results showed that Nrf2 binding to the ADAM10 promoter and HG interfered with this binding. Our data reinforce the notion that RAGE and Nrf2 might be sRAGE-regulating factors. Un der HG conditions, the treatment of EGCG reduced ROS generation and RAGE activation. EGCG-stimulated cRAGE release was likely caused by the upregulation of the Nrf2-ADAM10 pathway. EGCG inhibited HG-mediated NLRP3 inf lammasome activation at least partly by stimulating sRAGE, thereby reducing IL-1β release.
1. Introduction
Receptor for advanced glycation end products (RAGE) is a multiligand binding membrane receptor, which can bind to various damageassociated molecular patterns (DAMPs), such as advanced glycation end products (AGEs), amyloid-β protein, high mobility group box-1(HMGB1), and the S100/calgranulins family[1-2]. RAGE has been conf irmed by clinical studies to be closely related to the development of various chronic diseases, such as metabolic syndrome, diabetes mellitus, cardiovascular diseases[3], Alzheimer’s disease[4], cancer[5-6],and inflammaging[1]. More recently, the association of RAGE expression with morbidity and mortality in patients with COVID-19 has been highly concerning[7].
RAGE is a transmembrane protein composed of 404 amino acids. The main structure of RAGE consists of an extracellular immunoglobulin (Ig)-like domain composed of 344 amino acids at the N-terminal, a hydrophobic transmembrane-spanning region consisting of 19 amino acids, and an intracellular cytoplasmic tail composed of 43 amino acids at the C-terminal. The extracellular Ig-like domain of RAGE is composed of an IgV (V-type domain) followed by two IgCs (C-type domains) linked to it. The V-type domain is the binding site and is where RAGE recognizes ligands. After binding with ligands, the cytoplasmic tail of RAGE, which is highly charged, would form a complex with intracellular signal transduction molecules such as diaphanous-1 to facilitate the downstream signal transmission[2]. RAGE may be divided into 4 isoforms based on the structure, each with different physiological significance[8]: The first is full-length RAGE (fl-RAGE), which can recognize ligands and initiate intracellular signaling. The second is N-truncated RAGE, which lacks the extracellular IgV domain, and cannot transmit intracellular signals because it cannot recognize ligands. The third is C-terminal truncated RAGE, also known as endogenous secretory RAGE (esRAGE), which can perform cellular synthesis and secrete outside the cells. Although esRAGE can recognize ligands, it cannot transmit intracellular signals because of lacking a transmembrane domain and a cytoplasmic tail.The 4thtype is cleaved RAGE (cRAGE), which contains a complete extracellular IgV domain, but lacks a transmembrane domain and an intramembrane cytoplasmic tail. Usually, it is produced by fl-RAGE,which is cleaved by specific enzymes to release the extracellular IgV domain into blood circulation.
Soluble RAGE (sRAGE) is an isomer of RAGE. sRAGE may compete with RAGE for ligands because it contains a complete Ig-like IgV domain. In addition, as sRAGE lacks a transmembrane domain and a cytoplasmic tail, it cannot evoke RAGE-mediated signaling transduction after binding with specific ligands, and the complex would be free in plasma and excreted through urine. Therefore,sRAGE has been regarded as a scavenger of pro-inflammatory ligands[8]. Studies have shown plasma sRAGE levels closely related to diabetes mellitus, Alzheimer’s disease, and cardiovascular diseases[8-9].The administration of sRAGE to diabetic patients or diabetic mice inhibited RAGE activation and improved inflammation responses[9],indicating that the increase ofin vivosRAGE levels may be a research direction to prevent or treat diabetic complications.
esRAGE and cRAGE are collectively referred to as sRAGE,which is known to be released into the plasma via two mechanisms.The first is the spontaneous transformation from theRAGEgene through the post-transcriptional modification of the gene by alternative splicing, through which theRAGEgene is modified to produce esRAGE. The second pathway is the ectodomain shedding of the membrane RAGE proteins with metalloproteinases, through which cRAGE is released. Recent studies have demonstrated that a disintegrin and metallopeptidase 10 (ADAM10) are crucial enzymes determining cRAGE levelsin vivo[10-13]. Humans have around 600 proteases, 22 of which are members of the ADAM protease family[14].ADAM10 is expressed in multiple mammalian organs and has been linked to the progression of brain disorders, immunological dysfunction, chronic liver inflammation, and cancer in humans[15].Various mechanisms, including transcriptional and translational regulation, control ADAM10 activity. Several putative transcription factor binding sites are contained in the promoter region of ADAM10,which can drive its transcription. These include a retinoic acidresponsive element, SP1, and the spliced form of the X-box binding protein-1[16]. Patients with type 1 diabetes mellitus had elevated ADAM10 levels in circulation, which were positively associated with serum cRAGE levels[17]. On the contrary, ADAM10 expression in monocytes and sRAGE level in serum were reduced in type 2 diabetes mellitus patients with acute coronary syndrome[12]. These results revealed that ADAM10 is a crucial proteolytic enzyme responsible for shedding cRAGE; however, further mechanistic study on ADAM10 regulation under HG conditions is required.
The long-term hyperglycemia in people with diabetes would facilitate protein glycationin vivo, resulting in the accumulation of AGEs and the activation of RAGE, which would promote the generation of reactive oxygen species (ROS), resulting in oxidative stress and inflammatory reactions[5,18]. sRAGE is known to act as a decoy against DAMPs and block the inflammatory responses induced by RAGE, thus providing a preclinical biomarker of RAGE-mediated pathogenesis[2]. Therefore, exploring nutraceuticals that can promote the secretion of sRAGEin vivomay have a beneficial effect on improving the chronic inflammatory responses in diabetic patients.
Green tea is a common drink in daily life and is rich in polyphenolic antioxidants. Among them, catechins account for approximately 75%–80%, making up the highest proportion of polyphenols. Catechin may be divided into 4 types: epicatechin,epigallocatechin, epicatechin gallate, and epigallocatechin gallate(EGCG), among which EGCG has the highest antioxidant capacity due to the highest number of hydroxy groups[19-20]. Numerous studies have shown that EGCG exerted multiple physiological effects, such as hypoglycemic, antioxidant, anti-inflammatory, hypotensive,weight control, and blood lipid-lowering effects[19]. However, limited studies have explored whether EGCG promoted sRAGE secretion and its related molecular mechanisms. Therefore, a human monocyte culture model was adopted in this study to examine the effect of diabetes-relevant concentrations of glucose on sRAGE release and its possible mechanisms. Moreover, whether EGCG could attenuate the high glucose-induced inflammatory responses through stimulating sRAGE was further explored to serve as a pre-clinical reference for developing adjuvant therapies in diabetic patients.
2. Materials and methods
2.1 Chemicals
D-(+)-Glucose, 2’,7’-dichlorofluorescin diacetate (DCF-DA),dimethyl sulfoxide (DMSO), EGCG,N-acetyl-cysteine (NAC),anti-β-actin antibody, and protease inhibitor cocktail were purchased from Sigma Chemical (St. Louis, MO, USA). Antibodies to nuclear factor erythroid 2-related factor 2 (Nrf2) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). An anti-RAGE antibody was purchased from Abcam (Cambridge, MA, USA).Anti-ADAM10 antibody and sRAGE were purchased from R&D systems (Minneapolis, MN, USA). RAGE and Nrf2 siRNA were obtained from Tri-i biotech (Taipei, Taiwan, China). Fetal bovine serum (FBS) was purchased from Biological Industries (Panama,USA). RPMI 1640 medium (glucose-free), penicillin-streptomycin solution, sodium pyruvate, and UltraPure Agarose were obtained from Invitrogen (Carlsbad, CA, USA). All the chemicals and solvents used were of analytical grade.
2.2 Cell culture and treatments
The human THP-1 monocytic cell line (THP-1 cells) was obtained from the Bioresource Collection and Research Center (BCRC 60430, Food Industry Research and Development Institute, Hsin Chu,Taiwan, China), and cultured in RPMI glucose-free medium 1640 supplemented with 10% FBS, HEPES (10 mmol/L), 1% antibiotic solution (penicillin 100 U/mL and streptomycin 100 μg/mL),β-mercaptoethanol (50 μmol/L), and either 5.5 mmol/LD-glucose(normal glucose, NG) or 15 mmol/LD-glucose (high glucose, HG) in 5% CO2at 37 °C for 1–72 h. EGCG prepared in DMSO was added to cells with both the NG or HG media at the final concentrations of 5,10, and 20 μmol/L, respectively. Control cells received vehicle only,and the final concentration of DMSO was less than 0.05% of the total volume of tissue culture media.
2.3 Intracellular ROS production assay
Intracellular ROS generation was detected using a fluorescent probe, DCF-DA, as described previously[21]. At the end of incubation,cells (1 × 106cells/mL) were collected and resuspended in PBS.An aliquot of the suspension was loaded into a 96-well plate, and then DCF-DA was added (final concentration 10 μmol/L). The DCF fluorescence intensity was detected at different time intervals using a FLUOstar Galaxy Fluorescence Microplate Reader (BMG LABTECH, Offenburg, Germany) with an excitation wavelength of 485 nm and emission wavelength of 535 nm.
2.4 RNA preparation and reverse transcription-polymerase chain reaction (RT-PCR)
Total RNA was prepared from NG- or HG-treated THP-1 cells (1 × 106cells/mL) with a TRI Reagent™ Solution (Thermo Fisher Scientific Inc., Waltham, MA, USA) as described in the manufacturer’s manual. The primers used to amplify RAGE cDNA were forward (5’-CACCTTCTCCTGTAGCTTCA-3’)and reverse (5’-TGCCACAAGATGACCCCAA-3’); for S100A12 were forward (5’-ATGACAAAACTTGAAGAG-3’)and reverse (5’-CTACTCTTTGTGGGTGTG-3’); for ADAM10 were forward (5’-TTTGATGATGGCGTACTTGG-3’)and reverse (5’-AGTTTGTCCCCAGATGTTGC-3’); for those for 18S ribosomal RNA (18S rRNA) were forward(5’-TTGGAGGGCAAGTCTGGTG-3’) and reverse(5’-CCGCTCCCAAGATCCAACTA-3’). From each sample, cDNA corresponding to 500 ng–1 μg of RNA was reverse-transcribed,using RevertAidTMH Minus First Strand cDNA Synthesis Kit(Thermo Fisher Scientific Inc., Waltham, MA, USA) as described in the manufacturer’s manual. PCR analyses were performed on the cDNA preparations to detect RAGE, ADAM10, and 18S rRNA(as an internal standard) gene expression using a P×2 Thermal Cycler (Thermal Electron, Madison, WI, USA). The reactions were performed in a volume of 20 μL containing PCR Master Mix,template DNA (10 pg–1 μg), and indicated primers. After initial denaturation for 5 min at 95 °C, 25–35 cycles of amplification(the annealing temperature for RAGE, S100A12, ADAM10, and 18S rRNA was 56, 60, 60, and 59 °C, respectively) were performed,followed by 10 min of final extension at 72 °C. An aliquot from each PCR reaction was electrophoresed on a 1.0% agarose gel containing 0.005% SYBR®Safe. The gel was then photographed and quantification using BioDoc-It Imaging System (UVP, Upland, CA,USA). The results were expressed as fold stimulation over NG after normalizing the gene signal relative to the corresponding 18S rRNA from each sample.
2.5 Western blotting
The protein fractions were isolated from THP-1 cells cultured under NG or HG conditions with or without EGCG for 1–72 h.In gene silence assays, RAGE and Nrf2 siRNA plasmids were preincubated with THP-1 cells for 48 h using SureFECTTM transfection reagent (SABiosciences, Frederick, MD, USA) and incubated under HG conditions for a further 24 h. The cytosolic and nuclear fraction proteins by extracted by the Nuclear/Cytosol Fractionation Kit (Biovision, Mountain View, CA, USA). Protein concentration was measured by Bio-Rad Protein Assay Kit II(Hercules, CA, USA) using BSA as a standard. Total protein and compartmental protein extracts were separated on 5% SDS-PAGE for protein kinase C (PKC) detection and 12% SDS-PAGE for target proteins detection, then transferred to immobilon polyvinylidene difluoride membrane (Millipore) with transfer buffer composed of 25 mmol/L Tris-HCl (pH 8.9), 192 mmol/L glycine, and 20%methanol. The membrane was blocked in StartingBlockTMBlocking Buffers (Pierce, Rockford, IL, USA) and then incubated overnight at 4 °C with indicated primary antibodies (1:1 000 dilutions).After hybridization with primary antibodies, the membrane was washed with TBS containing Tween-20 three times, incubated with HRP-labeled secondary antibody for 60 min at room temperature, and washed with TBS containing Tween-20 three times. Final detection was performed with enhanced chemiluminescence detecting reagents(Millipore, Billerica, MA, USA). The relative expression of proteins was quantified densitometrically using Biospectrum AC Imaging System (UVP, Upland, CA, USA) and calculated according to the reference bands of loading control.
2.6 sRAGE, esRAGE, and IL-1β ELISA assays
THP-1 cells were treated with or without EGCG (5–20 μmol/L)under NG or HG conditions for 24–72 h. The supernatant was then harvested and assayed for sRAGE secretion using the Human RAGE DuoSet ELISA (DY1145, R&D systems Inc., Minneapolis, MN,USA), for esRAGE secretion using the esRAGE ELISA kit (K1009-1,B-Bridge International, Inc., San Jose, California, USA), and for IL-1β using READY-SET-GO! According to the manufacturer’s instructions, the human interleukin-1 beta ELISA kit (88-7010-22,eBioscience, San Diego, CA, USA). cRAGE was calculated by subtracting esRAGE from sRAGE. The pure medium (without cells)was incubated under the same conditions and was used as a blank control for the ELISA.
2.7 Chromatin immunoprecipitation (ChIP) assays
Following the manufacturer’s instructions and our previous study[21], ChIP assays were performed using the EZ-ChIPTM Chromatin Immunoprecipitation Kit (17-371, Millipore, Billerica,MA, USA). Briefly, THP-1 cells were cross-linked with 1%formaldehyde for 10 min, washed twice with cold PBS, resuspended in SDS lysis buffer (containing 1× Protease Inhibitor Cocktail II),and sheared with 4−5 sets of 10-s pulses on wet ice using a Misonix sonicator ultrasonic processor (model XL2020) set to 30% of maximum power. This was followed by centrifugation at 10 000 ×gfor 10 min to remove insoluble material. One-tenth of the total lysate was used for total genomic DNA as the “Input DNA” control. Dilution buffer (900 μL) containing Protease Inhibitor Cocktail II was added to each tube containing 100 μL of sheared, crosslinked chromatin. This was followed by immunoclearing with 60 μL of Protein G Agarose for 1 h at 4 °C. Immunoprecipitation was performed overnight at 4 °C with indicated antibodies. Precipitates were washed by resuspending the beads in 1 mL of each of the following cold buffers: Low Salt Immune Complex Wash Buffer, High Salt Immune Complex Wash Buffer, and LiCl Immune Complex Wash Buffer. Precipitates were washed twice with TE Buffer and extracted twice with 20% SDS containing 0.1 mol/L NaHCO3. Elutes were then pooled and heated at 45 °C for 4–5 h to reverse the crosslinks of the protein/DNA complexes to free the DNA. DNA fragments were purified with Spin Columns. For the PCR, 2 μL of the DNA sample was used. PCR primers correspond to sequences within the promoter regions: ADAM10, forward (50-cactcttccactccctcc-30) and reverse(50-agcctcaaacccttcctc-30).
2.8 Statistical analysis
The Statistical Package for the Social Sciences (SPSS)software version 12.0 was used for the analysis of statistical data.The experimental data were expressed as mean ± standard error(means ± SD), and one-way analysis of variance (ANOVA) was performed to analyze the differences within each group. Moreover,Duncan’s new multiple-range test was performed to analyze the differences between the mean values of each group. A statistical result ofP< 0.05 indicated a significant difference.
3. Results
3.1 HG stimulated ROS production and RAGE expression in monocytes
Compared with healthy subjects, diabetic patients suffer from higher oxidative stress burdens wing to long-term hyperglycemia[18,22].This study investigated whether ROS generation was affected in monocytes cultured in diabetes-relevant concentrations of glucose.A DCF-DA fluorescent probe was used to detect the levels of intracellular ROS production. The results showed that ROS generation was increased by approximately 130% in monocytes cultured in HG group for 24–72 h compared to that in the NG group (P< 0.05,Fig. 1A). Moreover, the expression ofRAGEmRNA in THP-1 cells was significantly higher when cultured in HG for 72 h than in the NG group by 131% (P< 0.05, Fig. 1B). Concomitantly, RAGE protein expression was upregulated by 127%, 135%, and 201%, respectively,when cultured in HG for 24, 48, and 72 h (Fig. 1C). These results suggested that high glucose concentrations induced oxidative stress and modulated RAGE activation. The HG condition was adopted in the study for the following investigation.

Fig. 1 Effect of normal and diabetes-relevant concentrations of glucose on ROS generation, RAGE expression, and sRAGE secretion in monocytes. (A) HG significantly increased intracellular ROS generation. HG induced mRNA transcription (B) and protein expression (C) of RAGE. (D) The secretion of sRAGE (D1),generated by cleavage (cRAGE) (D2) and mRNA splicing (esRAGE) (D3), were both inhibited by HG. (E) The treatment of sRAGE ablated RAGE activation in response to HG treatment. THP-1 cells were incubated with NG (normal glucose, 5.5 mmol/L) or HG (high glucose, 15 mmol/L) media for 24‒72 h with or without sRAGE (100 pg/mL). 18S rRNA was used as an internal control for gene expression. sRAGE and esRAGE levels in the cell-free supernatants were determined by ELISA. cRAGE was calculated by subtracting esRAGE from sRAGE. Results are means ± SD for n = 3. * and # mean significantly different from NG control group and HG-treated alone group at P < 0.05, respectively. The same below.
3.2 Monocytic sRAGE secretion was inhibited under HG conditions
sRAGE and esRAGE were measured using commercial ELISA kits. cRAGE was calculated by subtracting esRAGE from sRAGE.The results in Fig. 1D showed that the sRAGE secretion of monocytes cultured in HG was significantly lower than that of the NG group(P< 0.05). Compared with those in the NG group, sRAGE, esRAGE,and cRAGE secretion of monocytes cultured in HG for 72 h decreased by 33%, 31%, and 29%, respectively (Fig. 1D). These results indicated that HG was an influencing factor that inhibited sRAGE secretion. Of note, the addition of sRAGE (100 pg/mL) to HG media inhibited RAGE protein expression in monocytes (Fig. 1E), indicating the possibility of negative feedback between sRAGE and RAGE.
3.3 HG downregulated ADAM10 expression and counteracted by gene silencing of RAGE
Studies have confirmed ADAM10 to be a key enzyme for cellmediated cRAGE release[10-12]. Results in Fig. 2A showed that ADAM10 protein expression in monocytes was significantly inhibited after 12 h of culture in an HG condition (P< 0.05), which explained that the decrease of cRAGE release was related to the downregulation of ADAM10.
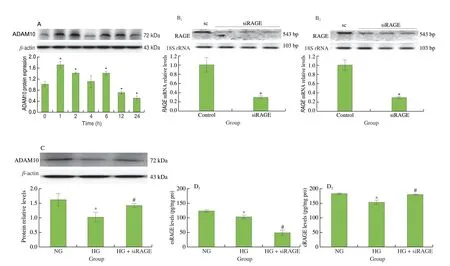
Fig. 2 Expressions of metalloprotease ADAM10 protein were downregulated by HG, and treatment of siRNA-RAGE counteracted HG-mediated inhibition on ADAM10 and cRAGE, but not esRAGE. (A) Modulatory effects of HG on ADAM10 protein expression. (B) Silenced RAGE gene expression in HG-treated cells.THP1 cells were transfected with RAGE siRNA for 48 h and then incubated in HG media for 12 h (B1) and 24 h (B2), respectively. 18S rRNA was used as an internal control for gene expression. (C) siRAGE counteracted HG-mediated inhibition on ADAM10 protein. (D) The opposite effects of siRAGE on esRAGE and cRAGE secretion in monocytes under HG conditions. Cells were incubated with NG (normal glucose, 5.5 mmol/L) or HG (high glucose, 15 mmol/L) media for 1–24 h. sc, scramble control.
Therefore, small interfering RNA (siRNA)-mediated gene silencing technologies were adopted to inhibit theRAGEmRNA in monocytes. THP-1 cells were transfected withRAGEsiRNA(30 nmol/L) for 48 h, and the inhibition rate of theRAGEgene was confirmed to reach over 72% at 24 h after transfection (Fig. 2B).Silencing theRAGEgene with RNAi alleviated the inhibition of ADAM10 expression (Fig. 2C) and cRAGE release (Fig. 2D) in monocytes under HG conditions, implying that HG may regulate ADAM10 expression by activating RAGE signaling, thereby inhibiting cRAGE release.
3.4 HG downregulated nuclear Nrf2 expression, and Nrf2 acted as an ADAM10 promotor regulating cRAGE release
After being translocated into the nucleus, the transcription factor Nrf2 is bound to the antioxidant response element (ARE) sequence and transcribed phase II detoxification and antioxidant enzymes[23]. In this study, it was found that nuclear Nrf2 protein levels significantly declined after 12 h of HG culture and that Nrf2 expression in the nucleus of the HG group was only 10% of that in the NG group(Fig. 3A), which indicated that HG might interfere with the nuclear translocation of Nrf2, thereby inhibiting the antioxidant enzymes and elevating the oxidative stress of cells.

Fig. 3 Nrf2 regulated ADAM10 gene translation in monocytes under HG conditions. (A) Nuclear expressions of the Nrf2 protein were regulated by HG in monocytes. (B) Silenced Nrf2 gene expression in HG-treated THP-1 monocytes. Cells were transfected with Nrf2-siRNA for 48 h and then incubated in HG media for 24 h. 18S rRNA was used as an internal control. Treatment of Nrf2-siRNA aggravated HG inhibition on ADAM10 expression (C) and cRAGE release (D).(E) The antioxidant response elements (ARE) sequence was found in the ADAM10 promoter region, as marked in blue (NCBI reference sequence:NT_010194.17). (F) ChIP assay for identifying the DNA-binding sites of Nrf2 protein in ADAM10 gene promotor in monocytes under HG conditions. Crosslinked chromatin samples were subjected to ChIP assays using antibodies to Nrf2. PCR was then performed with the immunoprecipitated DNA to amplify the ADAM10 promoter. sc, scramble control.
siRNA-mediated gene silencing technologies were further utilized. THP-1 monocytes were transfected with Nrf2 siRNA(30 nmol/L) for 48 h, and the inhibition rate of theNrf2gene reached 81% at 24 h after transfection (Fig. 3B). Notably, silencing of Nrf2 enhanced the reduction in ADAM10 protein expression (Fig. 3C)concomitant with reduced cRAGE levels (Fig. 3D) in response to HG treatment. This suggested that Nrf2 may be involved in the regulation of ADAM10 and thereby affected cRAGE release in monocytes.
Notably, the sequences in the promoter region of theADAM10gene were similar to ARE (5’-TGACXXXGC-3’, Fig. 3E). Therefore,a ChIP assay was performed to identify whether Nrf2 can bind to the binding site of the ADAM10 promoter and thus regulate ADAM10.Fig. 3F showed for the first time that Nrf2 could bind to the promotor region of ADAM10 regardless of whether the monocytes were cultured under NG or HG conditions. However, as HG inhibited the nuclear translocation of Nrf2 (Fig. 3A), the binding activity of Nrf2 to the ADAM10 promoter region was significantly lower than that in the NG group (Fig. 3F,P< 0.05).
3.5 EGCG reduced ROS production and RAGE activation and promoted cRAGE release under HG conditions
This study further investigated whether EGCG had a protective effect in response to HG. The results showed that EGCG at a concentration of 5–20 μmol/L minimized the increase in intracellular DCF-DA fluorescence intensities under HG conditions, indicating that EGCG had an antioxidant effect of reducing intracellular ROS generation (Fig. 4A). Given that sRAGE may act as a scavenger of proinflammatory ligands and competitively inhibit RAGE activation[8],Fig. 4A also showed that treatment of sRAGE reduces HG-induced oxidative stress in monocytes.

Fig. 4 EGCG counteracted HG-mediated negative aspects in monocytes. (A) Treatment of EGCG and sRAGE both reduced intracellular ROS generation under HG conditions. ROS was measured by DCF-DA probe as shown in green fluorescence microscopic images of treated cells. EGCG significantly inhibited RAGE and promoted ADAM10 protein expressions (B) and cRAGE release (C) at concentrations of 10 and 20 μmol/L. EGCG counteracted HG repression on Nrf2 nuclear translocation (D) and Nrf2 binding to the ADAM10 gene promotor (E). D1, D2: 4 h. D3, D4: 6 h. D5, D6: 12 h. Cells were incubated with NG (normal glucose, 5.5 mmol/L) or HG (high glucose, 15 mmol/L) media containing EGCG (5–20 μmol/L) or sRAGE (100 pg/mL) for 24 h.
EGCG inhibited HG-induced RAGE protein expression by 11%and 36% at 10 and 20 μmol/L, respectively (Fig. 4B). At the same concentration range, ADAM10 protein expressions were upregulated by EGCG treatment (Fig. 4B). Furthermore, EGCG stimulated cRAGE release by 22% and 41% at 10 and 20 μmol/L, respectively(P< 0.05, Fig. 4C). This suggested the positive effects of EGCG on neutralized RAGE activation and stimulated cRAGE release.
3.6 Mechanistic actions of EGCG in the release of cRAGE protein
In this study, ChIP confirmed that Nrf2 could bind to the ADAM10 promoter region and promote ADAM10 protein expression(Fig. 3F). The literature has pointed out that the metalloprotease ADAM10 is a crucial enzyme in cRAGE release. Therefore, it was inferred that Nrf2 might be a key factor regulating cRAGE release.The results in Fig. 4D showed that the nuclear expressions of Nrf2 in monocytes cultured under HG conditions were significantly lower than that in the NG group (P< 0.05). Intervention with EGCG(20 μmol/L) for 4, 6, and 12 h increased the nuclear expression of Nrf2 protein by 141%, 304%, and 576%, respectively. In addition, the ChIP results showed that EGCG (20 μmol/L) promoted the binding of Nrf2 to the ADAM10 promotor region (Fig. 4E), thereby upregulating ADAM10 protein expression (Fig. 4B). The above results showed that EGCG regulated the activity of downstream ADAM10 and promoted cRAGE release by promoting the nuclear translocation of Nrf2.
3.7 Anti-inflammatory effects of EGCG via attenuation of TXNIP-NLRP3 inflammasome activation under HG conditions
Recent studies revealed that proinflammatory cytokine IL-1β secretion in diabetic patients was related to NOD-like receptor protein 3(NLRP3) inflammasome activation[24]. Our data indicated that HG (25 mmol/L) promoted monocytic IL-1β secretion (P< 0.05,Fig. 5A), whereas intervention with NAC (antioxidant, 10 mmol/L),Z-YVAD (caspase-1 inhibitor, 10 μmol/L), RAGE siRNA, sRAGE(100 pg/mL), or EGCG (20 μmol/L) inhibited IL-1β secretion. This indicated that oxidative stress, inflammasome, and RAGE were involved in HG-induced IL-1β secretion, whereas intervention with sRAGE and EGCG inhibited these inflammatory responses (Fig. 5B).
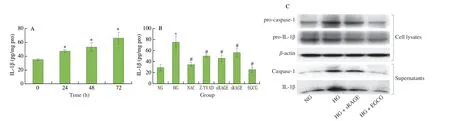
Fig. 5 EGCG negatively regulates IL-1β secretion through NLRP3 inflammasome inactivation under HG conditions. (A) HG progressively stimulated IL-1β secretion with incubation time (24–72 h). (B) HG-mediated IL-1β secretion was repressed by the treatment of antioxidant NAC (10 mmol/L), pan-caspase inhibitor Z-YVAD-FMK (100 μmol/L), RAGE-siRNA, sRAGE (100 pg/mL), and EGCG (20 μmol/L). Inhibitory effects of EGCG and sRAGE on caspase-1/IL-1β activation (C) and TXNIP protein expression (D) in HG-treated monocytes. (E) Co-IP demonstrated that the TXNIP-NLRP3 inflammasome protein complex levels were higher in the HG group than in the NG group. Treatment of EGCG or sRAGE inhibited TXNIP-NLRP3 binding complex formation. (F) HG may act as an NLRP3 inflammasome activator and the proposed mechanism of action. Cells were incubated with NG (normal glucose, 5.5 mmol/L) medium or HG (high glucose, 25 mmol/L) media for 0, 24, 48, and 72 h. IL-1β levels in cell-free supernatants were determined by ELISA.
Activated caspase-1 is known to cleave pro-IL-1β to mature IL-1β and is then released extracellularly together with IL-1β[25]. The analysis of HG media in monocytes in Fig. 5C showed that HG treatment promoted the release of caspase-1 and IL-1β in the media.In contrast, treatment with EGCG and sRAGE inhibited the activation of pro-caspase 1 to caspase-1 and reduced the levels of pro-IL-1β cleaved into mature IL-1β, which in turn inhibited IL-1β secretion (Fig. 5C).
When thioredoxin interaction protein (TXNIP) separates from thioredoxin (TRX), TXNIP can bind to NLRP3 and initiate inflammasome activation, promoting IL-1β release[25]. Fig. 5D shows that the treatment with EGCG and sRAGE reduced TXNIP protein expression by 37% and 17%, respectively. Co-immunoprecipitation analyses further demonstrated that the TXNIP-NLRP3 inflammasome protein complex formation occurred in HG treatment (Figs. 5E and F).EGCG and sRAGE substantially suppressed TXNIP-NLRP3 inflammasome complex formation and prevented TRX binding to TXNIP under HG conditions (Fig. 5E). In summary, EGCG may promote cRAGE release and inhibit the secretion of proinflammatory cytokine IL-1β by (1) inhibiting intracellular ROS generation to block RAGE and inflammasome activation and (2) promoting the binding of Nrf2 to the ADAM10 promoter region to enhance the enzyme activity of ADAM10 (Fig. 6).The stimulation of sRAGE release and the inhibition of NLRP3 inflammasome activation may reduce the inflammatory responses induced by hyperglycemia.
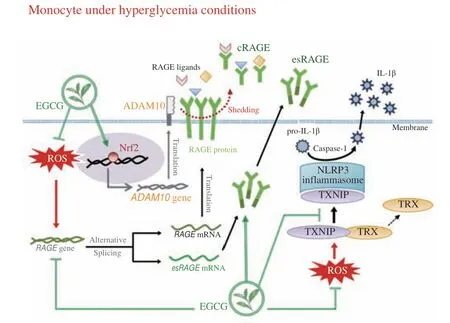
Fig. 6 Schematic illustration of the mechanisms of EGCG on sRAGE secretion and inflammasome activation under HG glucose conditions.
4. Discussion
Owing to the abnormal glucose metabolism in diabetics, the generation of free radicals may be accelerated, and antioxidants may be depleted in the body through the polyol pathway, hexosamine pathway, PKC activation, and AGEs generation, resulting in elevated oxidative stress[18]. For cells exposed to the HG conditions, RAGE is activated, and the expression of its ligands, such as S100A12 and HMGB1, may be promoted through ROS and AGEs[26-27]. S100A12,a pro-inflammatory protein secreted by neutrophils, is an endogenous RAGE ligand. Studies have shown that the S100A12 levels in diabetic patients were relatively high. Therefore, it has often been regarded as a physiological indicator of inflammation-related diseases[28-29]. In addition,the binding of AGEs to RAGE triggers signal transduction and stimulates a series of proinflammatory cytokine gene expressions, resulting in a feedback loop between RAGE and inflammatory responses[27].
In this study, monocytes were cultured in diabetes-relevant concentrations of glucose, and HG treatment induced considerable ROS production (Fig. 1A),S100A12gene expression (Fig. S1), and RAGE protein expression (Figs. 1B and C), which was in line with an earlier finding[30]. It was worth noting that HG treatment significantly reduced the secretion of monocytic sRAGE and that treatment with sRAGE antagonized the promotion of RAGE by HG (Figs. 1D and E).As sRAGE is an endogenous decoy, it can capture circulating pro-inflammatory ligands[8]. Therefore, clarifying the regulatory mechanisms of sRAGE may be an effective strategy to alleviate diabetic complications. Clinical studies have found the plasma levels of sRAGE in patients with diabetes, Alzheimer’s disease,and cardiovascular diseases to be lower than normal, indicating that sRAGE levels are closely related to these diseases[4,8-9].
sRAGE consists of two isoforms, esRAGE and cRAGE, which function as decoy receptors preventing the binding of fl-RAGE and ligands[8]. Studies have shown the metalloprotease ADAM10 to be the cleavage enzyme that plays a crucial role in cRAGE release[10-12]. We found that monocytes cultured in HG would lead to significant downregulation of ADAM10, decreasing esRAGE and cRAGE release (Fig. 2).However, RAGE siRNA-mediated gene silencing had different effects on these two sRAGE isoforms, and RAGE inhibition induced the release of cRAGE. These data suggested a possible negative regulatory mechanism between RAGE and ADAM10. We also observed an increase in the protein expression of ADAM10 during the first 2 h of monocyte culture in HG media (Fig. 2A), which may be related to PKC activation by HG treatment. Our previous study found that HG triggered PKC activation[31], whereas PKC has been shown to induce ADAM10 expression[32].
Devangelio et al.[33]indicated that the lower sRAGE levels in patients with type 2 diabetes were associated with their high oxidative stress burdens. This study also found thatRAGEgene silencing inhibited monocytic esRAGE release under HG conditions (Fig. 2D).In addition, the treatment of NAC, an antioxidant sulfhydryl substance and a glutathione precursor, counteracted the HG inhibition on esRAGE (Fig. S2), suggesting that ROS is likely to play an important role in the modulation of sRAGE secretion. The modulatory effect of HG on the transcription factor Nrf2 was further explored following these findings. As expected, HG caused transient activation of Nrf2 at the initial phase, all of which declined significantly beyond initial levels after 4 h of HG incubation (Fig. 3A). Similar observations were found in lipopolysaccharide-treated[34]and H2O2-treated cells[35]. Our data revealed that the activities of antioxidant enzymes in diabetic patients were lower than that in the normal subjects[36-37], which may be related to the HG-mediated reduction of Nrf2 activation[38].
Our findings also prove the first evidence thatNrf2gene silencing exacerbated HG inhibition on ADAM10 and cRAGE (Figs. 3C and D). ChIP results clearly demonstrated that Nrf2 binds to an ARE element in the ADAM10 promoter (Figs. 3E and F) and, in this action pathway, stimulates ADAM10 expression. In summary,nuclear localization of Nrf2 was ablated under HG conditions and subsequently inhibited ADAM10-mediated ectodomain shedding of extracellular RAGE, leading to cRAGE decline. Nrf2 is a redoxsensitive transcription factor that protects cells from toxic and oxidative stress by regulating the expression of drug detoxification and antioxidant stress response genes[39]. A growing body of research has emphasized the role of Nrf2 in defense against HG-induced oxidative damagein vitroandin vivo. Most recently, Albert-Garay and his colleagues[40]showed that HG reduces Nrf2 expression and Nrf2-regulated antioxidant enzyme expression, leading to alteration of the redox homeostasis in rat Müller retinal cells. A similar adverse effect in Nrf2-Keap1 was observed in the retina of donors with diabetic retinopathy[41].
EGCG is the most abundant green tea catechin. The phytotherapeutic actions of EGCG in disease management could be attributed to its antioxidant and anti-inflammatory properties[19-20].Our previous clinical studies have shown that cRAGE levels, rather than esRAGE levels, in circulation were increased in type 2 diabetic patients supplemented with EGCG-rich extracts[42]. This study further clarified that EGCG stimulates cRAGE release in monocytes via the modulation of RAGE and Nrf2 expressions, which in turn determines ADAM10-mediated ectodomain shedding of RAGE(Fig. 4). Although there is limited data on the crosstalk between Nrf2 and RAGE, the high-quality evidence that phytochemicals activate Nrf2 in human trials is insufficient[43]. The ROS generated through RAGE-ligand signaling might trigger a concomitant Nrf2 activation since evidence has shown that ROS activates Nrf2[39]. Otherwise, the elevated levels of Nrf2 stimulated by EGCG might also contribute to the inhibition of RAGE through the Nrf2-mediated ROS scavenging mechanisms. This postulate has consistent with Lee et al.[44], showing that cigarette smoke extracts alter RAGE expression via the activation of redox-sensitive DAMPs signaling through Nrf2 in human alveolar type II epithelial cells. However, more detailed studies are needed to determine their possible crosstalk.
Increasing evidence revealed that ROS acts as a kindling driving NLRP3 inflammasome activation[45]. Clinical trials have found that the circulation levels of proinflammatory cytokines, such as TNF-α,IL-1β, and IL-6, were higher than normal in glucose-intolerant and type 2 diabetic subjects[46]. Most of the various inflammatory diseases mediated by IL-1β are caused by the activation of the NLRP3/ASC/pro-caspase-1 inflammasome pathway. Zhou et al.[47]indicated that under hyperglycemia conditions, the disulfide bond between TRX and TXNIP dissociates and allows TXNIP to interact with NLRP3 inflammasome, leading to caspase-1 activation and IL-1β secretion. The results of this study also confirmed that the NLRP3 inflammasome pathway would be activated in monocytes cultured in HG conditions to promote IL-1β secretion (Fig. 5).
EGCG has been shown to exhibit antioxidant and anti-inflammatory properties, such as through the modulation of protective sRAGE and proinflammatory IL-1β secretion (Figs. 4 and 5). Tsai et al.[48]indicated that EGCG alleviated systemic lupus erythematosus by inhibiting the activation of the NLRP3 inflammasome complex and IL-1β production in mice. Cavet et al.[49]pointed out that EGCG inhibited ROS induced by glucose oxidase and reduced the release of IL-1β. In addition, EGCG reduced IL-1β-induced inflammatory responses by reducing mitogen-activated protein kinase phosphorylation as well as the transcriptional activity of NF-κB and AP-1. Our study reported that EGCG attenuated HG-triggered inflammasome activation by inhibiting the RAGE/ROS-TXNIPNLRP3 pathway. We proposed that the protective effect of EGCG, in addition to its antioxidant properties, may also be related to the action that it promoted sRAGE secretion since this study confirmed that NAC, sRAGE, and RAGE siRNA all had anti-inflammatory effects of inhibiting HG-mediated IL-1β secretion (Fig. 5B). Direct evidence from Co-IP further demonstrated that sRAGE kept the stability of the TXNIP-TRP complex, diminished TXNIP-NLRP3 binding, and abolished HG-induced increases in caspase-1 activity and IL-1β maturation.
Several literature reviews have indicated potential health risks associated with excessive green tea intake or high-dose EGCG supplements[50-52]. Specifically, phenolic hydroxyl groups at the B ring of EGCG are dehydrogenated in the presence of oxygen to formo-quinone derivatives, which may display prooxidant characteristics at high doses of EGCG[52]. Experimentsin vitroalso revealed that EGCG at concentrations of 10 mmol/L induced DNA strand breakage in whole blood lymphocytes[53]. Our data showed that no significantly increased in the DCF-DA fluorescence intensity was detected in EGCG-treated cells at a concentration range of 5–20 μmol/L(Fig. 4A), indicating the EGCG effects in this study may not contribute to its prooxidant properties. However, the effective dose of EGCG with minimum unfavorable effects should be clarified as an emerging issue in the future.
5. Conclusions
Tea catechin EGCG may reduce oxidative stress and inflammatory responses under HG conditions. The proposed mechanisms are represented in Fig. 6. The mechanisms by which EGCG promoted sRAGE release may be related to its inhibition of RAGE signaling activation and the promotion of the Nrf2-ADAM10 pathway. EGCG attenuates NLRP3 inflammasome activation and IL-1β release, at least in part, by promoting sRAGE. EGCG may have therapeutic potential in particulate hyperglycemia-associated inflammation in diabetics.
Conflicts of interest
The authors declare no conflict of interest.
Appendix A. Supplementary data
Supplementary data associated with this article can be found, in the online version, at http://doi.org/10.26599/FSHW.2022.9250129.

- 食品科学与人类健康(英文)的其它文章
- Betalains protect various body organs through antioxidant and anti-inf lammatory pathways
- Effects of Maillard reaction and its product AGEs on aging and age-related diseases
- Characterization of physicochemical and immunogenic properties of allergenic proteins altered by food processing: a review
- Polyphenol components in black chokeberry (Aronia melanocarpa)as clinically proven diseases control factors—an overview
- Food-derived protein hydrolysates and peptides: anxiolytic and antidepressant activities, characteristics, and mechanisms
- Recent advances in the study of epitopes, allergens and immunologic cross-reactivity of edible mango