
Procyanidin A1 and its digestive products alleviate acrylamide-induced IPEC-J2 cell damage through regulating Keap1/Nrf2 pathway
2024-01-24 01:11FngfngYnQunLuChengmingWngRuiLiu
食品科学与人类健康(英文) 2024年3期
Fngfng Yn, Qun Lu,b, Chengming Wng,b,, Rui Liu,b,c,
a College of Food Science and Technology, Huazhong Agricultural University, Wuhan 430070, China
b Key Laboratory of Environment Correlative Dietology (Huazhong Agricultural University), Ministry of Education, Wuhan 430070, China
c Key Laboratory of Urban Agriculture in Central China, Ministry of Agriculture and Rural Affairs, Wuhan 430070, China
Keywords: Procyanidin A1 Digestive products Acrylamide Nuclear factor erythroid 2-related factor 2 (Nrf2)Intestinal cell damage
ABSTRACT Our previous study has revealed that procyanidin A1 (A1) and its simulated digestive product (D-A1) can alleviate acrylamide (ACR)-induced intestine cell damage. However, the underlying mechanism remains unknown. In this study, we elucidated the molecular mechanism for A1 and D-A1 to alleviate ACR-stimulated IPEC-J2 cell damage. ACR slightly activated nuclear factor erythroid 2-related factor 2 (Nrf2) signaling and its target genes, but this activation could not reduce intestine cell damage. A1 and D-A1 could alleviate ACR-induced cell damage, but the effect was abrogated in cells transiently transfected with Nrf2 small interfering RNA (siRNA). Further investigation confirmed that A1 and D-A1 interacted with Kelch-like ECH-associated protein 1 (Keap1), which boosted the stabilization of Nrf2, subsequently promoted the translocation of Nrf2 into the nucleus, and further increased the expression of antioxidant proteins, thereby inhibiting glutathione (GSH) consumption, maintaining redox balance and eventually alleviating ACR-induced cell damage. Importantly, there was no difference between A1 and D-A1 treated groups, indicating that A1 can tolerate gastrointestinal digestion and may be a potential compound to limit the toxicity of ACR.
1. Introduction
Acrylamide (ACR) is a common contaminant generated during heat processing at high temperature in high-starch foods through the Maillard reaction[1]. ACR in food can be easily absorbed by the small intestine[2]. In the body, ACR and its main primary metabolite glycidamide can be further metabolized via additive reaction with glutathione (GSH), which usually causes the consumption of GSH[3]. GSH consumption tends to produce much reactive oxygen species (ROS). Increasing evidence has indicated that ACR-induced imbalance between the production and elimination of ROS will lead to apoptosis and oxidative stress damage in various organs and tissues such as the liver, kidney, brain, and ovary[4-6]. However, it has been largely ignored that ACR can also cause damage to the intestinal epithelial cells, the f irst barrier of defense against exogenous contaminants.
It has been reported that nuclear factor erythroid 2-related factor 2(Nrf2) is a crucial regulator of the defense mechanism against ACR-induced apoptosis and oxidative stress[7-8]. Under normal homeostasis, Nrf2 is anchored in the cytoplasm by Kelch-like ECH-associated protein 1 (Keap1) and then degraded. When cells suffer from stimuli, Nrf2 will escape from the Nrf2-Keap1 complex and be translocated into the nucleus, boosting the expression of antioxidant proteins[9]. ACR, as an electrophile, can have additive reaction with Keap1, thereby activating Nrf2 translocation into the nucleus and increasing the expression of antioxidant proteins[10].Although ACR-triggered Nrf2 activation has been found in some cells and tissues, there is no direct evidence for the interaction between ACR and Keap1[11]. Moreover, many researchers reported opposite results. Some researchers reported that ACR possesses a weak electrophilic ability, which could not lead to the translocation of Nrf2 into the nucleus. As a result, ACR-induced antioxidant protein expression was not detected[12]. However, some other researchers suggested that ACR could induce a decrease in the expression of antioxidant proteins[13-14]. Therefore, it is necessary to clarify the role of ACR in the Nrf2 pathway.
It has been reported thatN-acetylcysteine,Ganoderma atrumpolysaccharide, and crocin can suppress ACR-induced intestinal epithelial cell oxidative stress damage and apoptosis by decreasing the release of inflammatory factors and boosting the activity of antioxidant enzymes[4-6], but the molecular mechanism remains poorly understood. Rodriguez-Ramiro et al.[15]have indicated that procyanidins (PC) have certain protective effects against ACRinduced cytotoxicity in Caco-2 cells. Notably, gastrointestinal digestion may significantly decrease their bioactivities[16]. Fortunately,our previous studies have demonstrated that PC and their digestive products probably have equal inhibitory effects on ACR-induced cytotoxicity in IPEC-J2 cells, particularly procyanidin A1(A1) and its simulated digestive product (D-A1), which could effectively prevent ACR-induced GSH consumption and inhibit ACR-induced intestinal barrier dysfunction via the mitogen-activated protein kinase-mediated myosin light chain kinase pathway[17-18]. These findings indicate that A1may be a promising phytochemical that can endure gastrointestinal digestion to prevent ACR-induced small intestine epithelial damage.
In this study, we investigated the effect of ACR on Nrf2 signaling pathway in intestinal epithelial cells, aiming to providein vitromechanistic evidence for the inhibitory effect of A1and D-A1on ACR-induced cell damage through the Nrf2 pathway. Our findings may provide important implications for revealing the role of A1after digestion in alleviating ACR-induced small intestine epithelial cell damage.
2. Materials and methods
2.1 Materials and reagents
DMEM/F12 medium was obtained from HyClone (Logan,UT). Fetal bovine serum (FBS) was obtained from Gibco (Grand Island, USA). Penicillin/streptomycin, 0.25% trypsin and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide(MTT) were purchased from Sinopharm Co., Ltd. (Wuhan, China).ACR was obtained from Aladdin Co., Ltd. (Shanghai, China).JC-10 kit was purchased from KeyGen Biotech. (Jiangsu,China). Total superoxide dismutase (T-SOD), catalase (CAT),malondialdehyde (MDA), GSH, lactate dehydrogenase (LDH),Annexin-FITC/PI cell apoptosis detection, and bicinchoninic acid(BCA) protein assay kits were supplied by Nanjing Jiancheng Biology Engineering Co., Ltd. (Nanjing, China). 2’,7’-Dichlorofluorescin diacetate (DCFH-DA), nuclear and cytoplasmic protein extraction kit, cycloheximide, rabbit immunoglobulin G (IgG), and protein A/G magnetic beads were purchased from Beyotime Institute of Biotechnology (Jiangsu, China). RIPA lysis buffer, IP lysis buffer,chemiluminescence ECL assay kit, and horseradish peroxidase(HRP)-conjugated secondary antibodies were purchased from Servicebio Co., Ltd. (Wuhan, China). The primary antibodyβ-actin was obtained from Ruiying Biological Co., Ltd. (Suzhou, China). The primary antibody against histone H3 was from Proteintech Group, Inc.(Wuhan, China). The primary antibody against Nrf2 was purchased from Cell Signaling Technology Co. (Danvers, MA, USA). The primary antibodies against Keap1, glutamate-cysteine ligase catalytic(GCLC), glutamate-cysteine ligase modifier subunit (GCLM), heme oxygenase 1 (HO-1), and NADPH:quinone oxidoreductase (NQO1)were provided by Beijing Boiss Biotechnology Co. Ltd. (Beijing,China). Lipofectamine 2000 was purchased from Invitrogen (CA,USA). Negative control small interfering RNA (NC siRNA) and Nrf2 siRNA were synthesized by GenePharma Co., Ltd. (Shanghai, China).
2.2 Preparations of A1 and D-A1
A1was isolated from peanut skin in our laboratory, and its purity was higher than 98% as analyzed by HPLC. D-A1was prepared with the method reported in our previous study[18]. Briefly, A1was conducted a simulated salivary, gastric and intestinal digestion. Then, the samples were centrifuged to obtain the supernatant which finally was freeze-dried to obtain D-A1. D-A1was mainly composed of three different procyanidin A-type dimers through HPLC-Q-TOF-MS2analysis.
2.3 Cell culture
IPEC-J2 cells were obtained from the Cell Bank of Type Culture Collection of Chinese Academy of Sciences (Shanghai, China), and cultured in DMEMF/12 medium supplemented with 10% FBS and 1%penicillin/streptomycin under a humidified atmosphere of 5% CO2at 37 °C. According to our previous results, cells were seeded and pretreated with A1or D-A1(0.2, 1, or 2 μmol/L) for 4 h, followed by treatment with ACR (7.5 mmol/L) for 4–20 h in this study[18].
2.4 Determination of LDH release
IPEC-J2 cells were grown in 6-well plates (3 × 105cells/well).Cells were incubated with A1or D-A1for 4 h, and then treated with ACR for 20 h. The supernatant was collected. The level of LDH release was detected according to the manufacturer’s instruction and expressed as units per liter supernatant (U/L).
2.5 Mitochondrial membrane potential (MMP) and cell apoptosis assays
IPEC-J2 cells were seeded in 35 mm glass dishes (3 × 105cells/well)and incubated with A1or D-A1for 4 h and ACR for another 4 or 20 h. Cells were stained with JC-10 or Annexin-V-FITC/PI dye solutions for 15 min in the dark. Then, cells were washed twice and photographed using a confocal laser scanning microscope (CLSM,Olympus FLUOVIEW FV3000, Tokyo, Japan) at 490 nm excitation and 520 or 590 nm emission wavelengths to measure the green and red fluorescence, respectively. The fluorescence intensity was calculated by the ImageJ software.
2.6 Measurement of MDA, T-SOD, CAT, and GSH contents
IPEC-J2 cells were seeded in 6-well plates (3 × 105cells/well).Cells were incubated with A1or D-A1for 4 h, and then treated with ACR for 20 h. The following procedures were carried out according to the manufacturer’s instructions. The MDA content was expressed as nanomoles per microgram protein (nmol/μg prot). The T-SOD and CAT activities were expressed as units per milligram protein (U/mg prot),and GSH level was expressed as micromoles per gram protein(μmol/g prot).
2.7 Determination of intracellular ROS
The fluorescent probe DCFH-DA was used to detect the levels of intracellular ROS. IPEC-J2 cells (8 × 103cells/well in 96-well black plates or 3 × 105cells/well in 35 mm glass dishes) were incubated with A1or D-A1for 4 h followed by ACR incubation for 6 h. The cells were incubated with 10 μmol/L DCFH-DA for 30 min.Finally, cells were washed twice with PBS, and the fluorescence intensity was measured using a Synergy HTX multi-mode reader(Bio-Tek, Winooski, VT) or an Olympus FLUOVIEW FV3000 microscope (Tokyo, Japan) at 485 nm excitation and 525 nm emission wavelengths.
2.8 siRNA transfection
siRNA transfection was conducted as described previously[19].IPEC-J2 cells were cultured until 50% confluence and transfected with Nrf2 siRNA or NC siRNA using Lipofectamine 2000. After 24 h of transfection, Western blot was performed to test the Nrf2 expression. For cell viability and intracellular ROS detection,cells were treated with A1or D-A1preceding ACR incubation.The siRNA sequences targeting pig Nrf2 were as follows:sense 5’-GCCCAUUGAUCUCUCUGAUTT-3’ and antisense 5’-AUCAGAGAGAUCAAUGGGCTT-3’.
2.9 Cell viability test
Cell viability was indirectly evaluated by MTT assay. After treatment, the cells were treated with MTT solution (0.5 mg/mL) for 4 h at 37 °C. The supernatant was removed and 150 μL DMSO was added to each well. Finally, the absorbance was measured at 490 nm with a microplate reader (Multiskan GO, Thermo Fisher, USA). The viability of cells was quantified as a percentage compared to the blank treatment.
2.10 Extraction of nuclear and cytoplasmic proteins
IPEC-J2 cells (1 × 106cells/well) were cultured in 60 mm dishes for 24 h. After treatment with A1or D-A1for 4 h, the cells were exposed to ACR for 4 h. Nuclear and cytoplasmic proteins were separated by nuclear and cytoplasmic protein extraction kits according to the manufacturer’s instructions. Then, the proteins were conduct Western blot analysis.
2.11 Stabilization of Nrf2
The stabilization of Nrf2 was analyzed by protein decay experiments. IPEC-J2 cells were seeded in 6-well plates(3 × 105cells/well) and treated with A1or D-A1for 4 h. After treatment with 50 μmol/L cycloheximide, the cells were collected at 0, 1, 2, and 3 h.Then, cells were collected to conduct western blot analysis.
2.12 Co-immunoprecipitation
After treatment with A1or D-A1for 4 h and ACR for 2 h, the cells were lysed with IP lysis buffer and centrifuged at 12 000 ×gfor 15 min at 4 °C to collect the supernatant. The protein lysates(500 µg) were incubated with 1 µg normal rabbit IgG and 15 μL of the protein A/G magnetic beads at room temperature for 60 min,and then the supernatant was collected by magnetic frame. The supernatant was incubated with 3 μg anti-Nrf2 or 3 μg normal rabbit IgG at 4 °C overnight and then incubated with 20 μL the protein A/G magnetic beads at 4 °C for 2 h. The immunoprecipitation complex was separated by a magnetic frame to collect the beads,and the beads were washed three times with IP lysis buffer. The complexes were eluted with 100 μL loading buffer, boiled at 100 °C for 5 min, and analyzed by Western blotting.
2.13 Western blot
After the indicated treatments, cells were lysed by RIPA lysis buffer and the total protein content was measured with a BCA protein assay kit. Twenty micrograms of protein was isolated by 8%–12%SDS-PAGE and subsequently transferred to 0.22 μm polyvinylidene fluoride membranes. Then, the membrane was blocked with 5%non-fat milk for 2 h at room temperature and incubated with primary antibodies against Nrf2 (1:1 000), Keap1 (1:500), GCLC(1:500), GCLM (1:500), NQO1 (1:500), HO-1 (1:500), histone H3(1:2 000) andβ-actin (1:2 000) overnight at 4 °C. After incubation with HRP-conjugated secondary antibody at room temperature, the immunoreactive proteins were visualized by a chemiluminescence ECL assay kit. The intensity of each band was quantified by ImageJ software and normalized againstβ-actin or histone H3.
2.14 Molecular docking
Molecular docking assay for the interaction between A1and Keap1 protein was performed using the AutoDock Vina. The 3D structure of A1was generated and energy-minimized using ChemDraw software. The crystal structure of the Keap1 protein (PDB ID: 4L7B) was obtained from the Protein Data Bank (www.pdb.org). Before docking, all water molecules and ligands were removed from the protein, and then the protein was added with hydrogen. The grid box for the Keap1 was fixed at 2.914, –3.687, –25.887 forX,Y,Zcoordinates, respectively. All other docking parameters were default unless otherwise stated. The best-scoring pose as judged by the Vina docking score was selected and visually generated using the PyMOL software.
2.15 Statistical analysis
All data analyses were conducted using the SPSS 24.0 software (SPSS Inc., Chicago, IL, USA). Data represent mean ±standard error of the mean (SEM) of three or more replicates.One-way analysis of variance (ANOVA) followed by Duncan’s test was performed to identify the differences among multiple groups. Differences atP< 0.05 were considered as statistically significant. Graph plotting was performed with GraphPad Prism 8.0(CA, USA).
3. Results
3.1 A1 and D-A1 attenuate ACR-induced IPEC-J2 cell apoptosis
The activity of extracellular LDH, a key indicator of cell death, showed obvious decreases after pretreatment with different concentrations of A1or D-A1before ACR exposure (Fig. 1A). MMP is an important factor for maintaining normal mitochondrial function and cellular activity. A decrease in MMP also reflects a state of early apoptosis[20]. After 4 h of ACR treatment, MMP showed a sharp decline, while A1and D-A1resulted in an evident suppression of ACR-induced MMP loss (Figs. 1C, D). As expected, the Annexin V-FITC/PI staining results confirmed the protective effect of A1and D-A1against ACR-induced cell apoptosis (Fig. 1B). These results demonstrated that A and D-A1are able to attenuate ACR-induced IPEC-J2 cell damage.
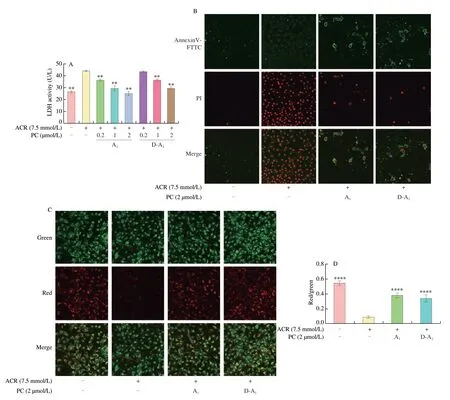
Fig. 1 A1 and D-A1 protect IPEC-J2 cells against ACR-induced cell damage. (A, B) IPEC-J2 cells pretreated with A1 or D-A1 (0.2, 1 or 2 μmol/L) for 4 h and then exposed to ACR (7.5 mmol/L) for 20 h. (A) LDH release in the medium was detected (n = 3). (B) Annexin V-FITC/PI staining was used to analyze cell apoptosis, which was observed under CLSM (magnification 40×). (C, D) Cells exposed to ACR (7.5 mmol/L) for 4 h after A1 or D-A1 (2 μmol/L) treatment with 4 h, the MMP was determined using JC-10 probe. (C) Cells were photographed using CLSM (magnification 40×). (D) Red fluorescence density ratio to green fluorescence density (n = 6). The results are represented as mean ± SEM. Significant at *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.000 1, in comparison with ACR treatment alone.
3.2 A1 and D-A1 inhibit ACR-induced oxidative stress in IPEC-J2 cells
GSH is essential for the intracellular detoxification of ACR[1].In this study, A1and D-A1were found to reduce ACR-induced GSH consumption in a concentration dependent manner (Fig. 2A).Consistently, A1and D-A1pretreatment remarkably increased the activities of CAT and T-SOD relative to ACR alone (Figs. 2B, C).Since the consumption of antioxidant substances might cause cell oxidative stress damage, we examined the role of A1and D-A1in alleviating this cell damage. As expected, intracellular ROS generation and MDA formation could be inhibited by A1or D-A1pretreatment (Figs. 2D–F).
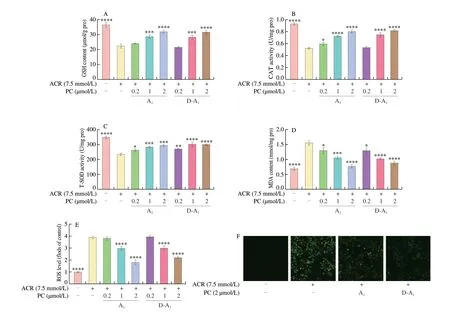
Fig. 2 A1 and D-A1 inhibits ACR-induced oxidative stress damage in IPEC-J2 cells. (A–D) IPEC-J2 cells were treated with A1 or D-A1 (0.2, 1, or 2 μmol/L) for 4 h before challenging by ACR (7.5 mmol/L) for 20 h. Levels of (A) GSH, (B) CAT, (C) T-SOD, and (D) MDA in cells (n = 3). (E, F) Cells were preincubated with A1 or D-A1 (0.2, 1, or 2 μmol/L) for 4 h before exposure to ACR (7.5 mmol/L) for 6 h. (E) The intracellular ROS level was measured by fluorescence microplate(n = 4). (F) The intracellular ROS level was observed under a CLSM (magnification 40×). The results are represented as mean ± SEM. Significant at *P < 0.05,**P < 0.01, ***P < 0.001, and ****P < 0.000 1, in comparison with ACR treatment alone.
3.3 A1 and D-A1 elicit the expression of anti-oxidative proteins
The synthesis of antioxidant substances (including GSH) is regulated by antioxidant reaction elements (ARE)[21]. We then determined the expression of GCLC, GCLM, HO-1, and NQO1 proteins, which are regulated by ARE. Surprisingly, ACR triggered the expression of HO-1 and NQO1 proteins, and A1or D-A1pretreatment further boosted the expression levels of these proteins (Figs. 3A, B).Interestingly, ACR alone could not promote the protein expression of GCLC and GCLM, whereas both A1and D-A1pretreatment could increase their expression (Figs. 3C, D). Given the potential regulatory effect of Nrf2 on ARE, we investigated the expression of Nrf2 protein. As a result, A1or D-A1dramatically impeded ACR-induced decrease in Nrf2 protein (Fig. 3E). These results demonstrated that A1and D-A1probably activate the expression of antioxidant proteins through Nrf2 signaling pathway.
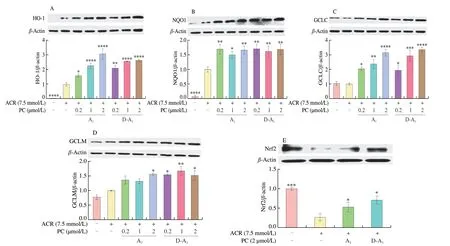
Fig. 3 A1 and D-A1 elicit the expression of anti-oxidative proteins in IPEC-J2 cells. (A–E) Cells were pretreated with A1 or D-A1 (0.2, 1, or 2 μmo/L) for 4 h, followed by treatment with ACR (7.5 mmol/L) for an additional 20 h. Protein levels of (A) HO-1, (B) NQO1, (C) GCLC, (D) GCLM, and (E) Nrf2 were determined by Western blot, β-actin was used to normalize the expression of these proteins (n = 3). The results are represented as mean ± SEM. Significant at*P < 0.05, **P < 0.01 ***P < 0.001, and ****P < 0.000 1, in comparison with ACR treatment alone.
3.4 Nrf2 knockdown blunts the protective effect of A1 and D-A1
To verify the participation of Nrf2 in the inhibitory effect of A1and D-A1on ACR-induced IPEC-J2 cell damage, we transfected the IPEC-J2 cells with siRNA to inhibit the expression of Nrf2. In Nrf2 siRNA transfected cells, the expression of Nrf2 protein was reduced by nearly 70%, indicating successful knockdown of Nrf2(Fig. 4A). Compared with NC siRAN treatment, Nrf2 siRNA treatment promoted ACR-induced the increase in the level of ROS(Fig. 4B) and the reduction of cell viability (Fig. 4C), indicating that Nrf2 is an important regulator of the toxicity of ACR. Besides,after Nrf2 siRNA treatment, A1and D-A1partially lost their abilities to scavenge ROS and the cell viability increased in ACR-induced IPEC-J2 cells, compared with NC siRAN treatment. Therefore, it could be inferred that the protective effect of A1and D-A1against ACR-induced IPEC-J2 cell damage is dependent on the activation of Nrf2/ARE signaling.

Fig. 4 Knockdown of Nrf2 diminishes the protective effects of A1 and D-A1. (A) Cells were treated with Nrf2 siRNA or NC siRNA for 24 h. Protein expression of Nrf2 was quantified using Western blot, β-actin was used to normalize the expression of the protein (n = 3). (B) After transfection with Nrf2 siRNA or NC siRNA for 24 h, the cells were incubated with A1 or D-A1 (2 µmol/L) for 4 h before treatment with ACR (7.5 mmol/L) for additional 6 h. The intracellular ROS level was measured by fluorescence microplate (n = 4). (C) After transfection with Nrf2 siRNA or NC siRNA, cells were pretreated with A1 or D-A1 (2 µmol/L) for 4 h, followed by incubation with ACR (7.5 mmol/L) for 20 h, and cell viability was measured by MTT (n = 6). The results are represented as mean ± SEM. Significant at ***P < 0.001 in comparison with control group; ns, not significant (P > 0.05), significant at ##P < 0.01, and ####P < 0.000 1 in comparison with the indicated group.

Fig. 5 A1 and D-A1 activate the Keap1/Nrf2 signaling pathway via promoting Nrf2 translocation into the nucleus and enhancing Nrf2 stabilization.(A–C) IPEC-J2 cells were treated with A1 or D-A1 (2 µmol/L) for 4 h and exposed to ACR (7.5 mmol/L) for another 4 h. The nuclear and cytoplasmic cell proteins were separated and subjected to Western blot analysis. Histone H3 and β-actin were used to normalize the expression of nuclear and cytoplasmic proteins,respectively (n = 3). (A) Images of Western blot bands. Expression of (B) cytoplasmic Nrf2 and (C) nuclear Nrf2 proteins. (D, E) IPEC-J2 cells treated(D1) without or with (D2) A1 or (D3) D-A1 (2 μmol/L) for 4 h. Cycloheximide (50 μmol/L) was added to block protein synthesis (0, 1, 2, 3 h). Total cell lysates were conducted for Western blot analysis, β-actin was used to normalize the expression of the protein (n = 3). (D) Images of Western blot bands. (E) Histogram of the expression of Nrf2 protein in IPEC-J2 cells. (F,H) Cells pretreated with A1 or D-A1 (2 µmol/L) for 4 h, followed by treatment with ACR (7.5 mmol/L) for additional 20 h. Protein levels were determined by Western blot, β-actin was used to normalize the expression of these proteins (n = 3). (F) Images of Western blot bands. Expression of (G) Keap1 and (H) Keap1 dimer proteins. The results are represented as mean ± SEM. Significant at *P < 0.05, **P < 0.01, and ***P < 0.001 in comparison with ACR treatment alone.
3.5 A1 and D-A1 activate the Nrf2 signaling pathway in ACRinduced IPEC-J2 cells
Nrf2 stabilization in the cytoplasm and accumulation in the nucleus are necessary for the activation of ARE[21]. The Western blot results indicated that ACR could slightly promote the translocation of Nrf2 into the nucleus, which was further enhanced by A1or D-A1pretreatment (Figs. 5A-C). Furthermore, we investigated the capacity of A1and D-A1to stabilize the Nrf2 protein. The results revealed that A1or D-A1treatment could increase the stability of the Nrf2 protein(Figs. 5D and E). A1and D-A1treatment reduced the expression of Keap1 protein though it did not promote the formation of Keap1 dimer, which may facilitate the release of Nrf2 from Nrf2-Keap1 complex (Figs. 5F–H). These results suggested that A1and D-A1activate the Nrf2 signaling pathway by stabilizing Nrf2 and facilitate the subsequent translocation of Nrf2 to the nucleus.
3.6 A1 and D-A1 interfere with the interaction between Keap1 and Nrf2
To validate that A1and D-A1can boost the release of Nrf2 from the Keap1-Nrf2 complex, a co-immunoprecipitation analysis was conducted. When Nrf2 was immunoprecipitated with its primary antibody, the level of Keap1 decreased in the cells exposed to ACR alone, which was further aggravated by A1and D-A1pretreatment(Figs. 6A and B). These results suggested that A1and D-A1could break the formation of the Keap1-Nrf2 complex. Our previous study has demonstrated that D-A1is mainly composed of three procyanidin A-type dimers, and A1is the dominant component[18]. Therefore, we investigated the possible binding mode between A1and Keap1 by molecular docking. The results revealed that the binding energy of A1with Keap1 was –10.3 kcal/mol. The hydrogen atom of phenolic hydroxyl in A1formed hydrogen bonds with Ile 559, Val 606, Val 561, Val 514, and Asn 517 (Fig. 6C). A π-π interaction was detected between Val 467 and a benzene ring of A1(Fig. 6D). Therefore, these interactions may enhance the binding between A1and Keap1. These findings indicated that A1and D-A1could directly bind with Keap1,thereby disrupting the interaction between Keap1 and Nrf2.
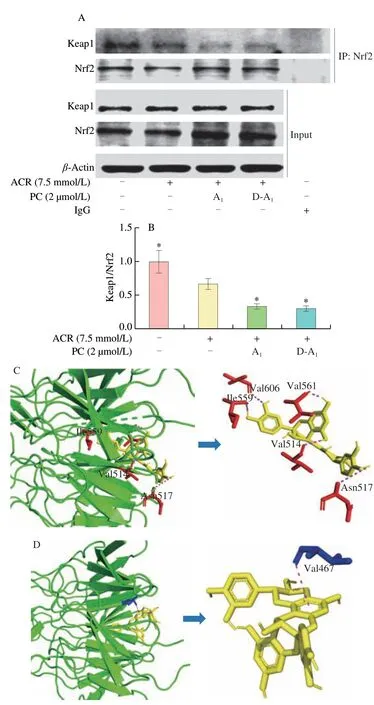
Fig. 6 A1 and D-A1 potentially disrupt the Nrf2-Keap1 interaction.(A, B) IPEC-J2 cells were incubated with A1 or D-A1 (2 μmol/L) for 4 h and then treated with ACR (7.5 mnol/L) for 2 h. Interaction of Keap1 with Nrf2 was visualized using co-immunoprecipitation assay. The IgG was used as a negative control (n = 3). (A) Immunoprecipitation assay for Nrf2.(B) Quantification of Keap1 binding with Nrf2. (C, D) Molecule docking of the interaction between A1 and Keap1. (C) Interactions between amino acid residues and A1 through hydrogen bonds. (D) Interactions between amino acid residues and A1 through π-π interactions. The results are represented as mean ±SEM. Significant at *P < 0.05 in comparison with ACR treatment alone.
4. Discussion
ACR is an exogenous contaminant produced during the processing of high-carbohydrate foods at temperature greater than 120 °C. There has been increasing research on its renal toxicity,liver toxicity, reproductive toxicity, and neurotoxicity[1]. The small intestine is the main absorption organ and the first line of defense against ACR, but the toxicity of ACR to the small intestine itself is usually ignored. ACR is generally metabolized through two pathways. On the one hand, ACR is metabolized to glycidamide by cytochrome P450 enzyme[22], and then glycidamide can be further converted into non-toxic glyceramide by epoxide hydrolase[23]. On the other hand, ACR or glycidamide is coupled with GSH by glutathioneS-transferase to produce GSH conjugates, which are then rapidly transformed into mercapturic acid metabolites[1]. Therefore, GSH may be a key substance to suppress the toxicity of ACR. Consistent with our previous research, the results of the present study demonstrated that ACR increased the consumption of GSH in IPEC-J2 cells, which could be reversed by A1and D-A1[18]. ACR-induced GSH consumption would cause ROS accumulation. Moreover, as an electron-deficient electrophile, ACR easily reacts with proteins and molecular structures with nucleophiles and electron-dense molecules (such as –OH, –SH,–NH2functional groups), and then produces ROS[8,24].
Excessive ROS production accompanied by cell membrane phospholipid damage would induce lipid peroxidation and diminish antioxidant enzyme activity (such as T-SOD and CAT), thereby disrupting cell redox balance, and eventually resulting in cell damage. To maintain a normal oxidation-reduction state, Nrf2/ARE,as a master switch for the transcription of antioxidant genes, may be a promising target that activates the detoxification response of phytochemicals (such as carnosic acid, allicin and melatonin) to ACR[7,25-26]. Here, we also found that Nrf2 knockdown increased the toxicity of ACR, implying that Nrf2 is a pivotal factor to the toxicity of ACR. Under basal conditions, Nrf2 is bound with Keap1,a substrate adaptor protein for the Cullin3-containing E3-ligase complex, which promotes Nrf2 ubiquitination and its subsequent degradation[27]. Upon stimulation, this binding is broken, leading to the release and translocation of Nrf2 into the nucleus, and further activating the transcription of antioxidant genes[28]. There has been some controversy about whether ACR can activate the Nrf2/ARE pathway. Theoretically, ACR, as a soft electrophile withα,β-unsaturated carbonyl structure, is easy to form adducts with soft nucleophilic Keap1 cysteine residues, such as Cys 257, Cys 273, Cys 288, and Cys 297[29]. Conversely, some other researchers reported that ACR has relatively weak electrophilicity and the nucleophilicity of Keap1 cysteine residues is lower than expected[12]. Indeed, the adducts of ACR and Keap1 were not detected in ACR-induced rat brains by mass spectrometric analysis[29]. Some studies have also confirmed that ACR administration is insufficient to stimulate Nrf2 nuclear translocation, which further prevents the activation of ARE target genes[11,30]. Surprisingly, there has been increasing evidence indicating that under long-term and ongoing ACR stimulation, Nrf2 is released and translocated into the nucleus, where it activates the expression of antioxidant-associated genes, affecting the adaptation of tissues/cells to stress and restoring a new homeostatic state[31]. However,there has been no direct evidence that ACR can promote Nrf2 release by combining with Keap1[8,32]. In this study, we verified that ACR could slightly promote the translocation of Nrf2 into the nucleus and subsequently increase the expression of HO-1 and NQO1 proteins.Importantly, we revealed that ACR could facilitate the release of Nrf2 by inhibiting the binding of Keap1 and Nrf2. Although ACR exposure activated the Nrf2/ARE pathway, cell apoptosis and oxidative stress damage were still markedly increased in ACR-treated IPEC-J2 cells,probably because ACR failed to activate the expression of GCLC and GCLM and only slightly promoted that of HO-1 and NQO1, and as a result the expression of antioxidant proteins was insufficient to rescue the cells. Therefore, many studies have proposed that additional activation of Nrf2-downstream antioxidant proteins may be an ideal therapy to inhibit ACR-induced cell damage[26,33]. Previous studies have reported that some natural compounds such as carnosic acid,lipoic acid and silymarin are specific Nrf2 activators and can provide protection against ACR-induced cytotoxicity[26,34-35]. These results suggest that phytochemical-triggered Nrf2 activation may depend on the Keap1-dependent pathway in ACR-induced cells. However, it is a pity that this special activation mechanism has only be indirectly verified and remains largely unknown.
PC is a class of natural dietary polyphenols with the capability of ameliorating inflammation, endoplasmic reticulum stress, oxidative stress damage and apoptosis because they can directly scavenge free radicals and activate Nrf2/ARE signaling[36]. Our previous studies have demonstrated that PC may be an excellent inhibitor of ACR-induced intestinal epithelial cell damage. Among several PC and their digestive products, A1and D-A1possess the optimal protective effect[18]. Here, by knockdown of Nrf2 expression, we demonstrated that Nrf2 activation mediates the protective effect of A1and D-A1against ACR-induced cell damage. A1and D-A1treatment significantly promoted the translocation of Nrf2 into the nucleus,which signifies its potential to activate ARE. This promotion effect was also affirmed by increases in the levels of its downstream antioxidative proteins, particularly GCLC and GLCM, which are closely related to GSH synthesis[37]. There may be two main reasons for polyphenols to contribute to the Nrf2 nuclear translocation. Firstly,polyphenols might promote the synthesis of Nrf2, and the newly synthesized Nrf2 may escape from the Keap1 binding and be further translocated into the nucleus. Secondly, polyphenols can disrupt the Nrf2-Keap1 complex, leading to the release and translocation of Nrf2 into the nucleus[38]. In this study, A1and D-A1pretreatment increased the expression of Nrf2 and decreased that of Keap1, which may contribute to the release and nuclear translocation of Nrf2.However, the most effective and fastest way to activate Nrf2 is still to destroy the interaction between Nrf2 and Keap1[39]. The known Nrf2 activators such as itaconic acid, sulforaphane andtert-butyl hydroquinone can directly form covalent adducts with the cysteine sulfhydryl groups of Keap1 protein to prevent the degradation of Nrf2[39-40]. Different from the above activators, A1docks to Keap1 through specific hydrogen bonds and hydrophobic interactions to disrupt the interaction between Keap1 and Nrf2 and prevent Nrf2 degradation. Moreover, several upstream kinases mediated by PC could phosphorylate Nrf2 at specific serine and/or tyrosine residues to stabilize Nrf2, which is another important way to activate antioxidant proteins[41-42]. However, further investigations are still required to fully elucidate the detailed molecular mechanism underlying the activation of the Nrf2/ARE signaling pathway by PC.
Although the biological properties of PC have been extensively studied, their activities after absorption and digestionin vivoremain poorly understood. In particular, it is difficult to quantify and detect most metabolites of PC, resulting in great difficulty in explaining the potential activity mechanism of PC. In this study, no marked difference was observed in the inhibitory effect and molecular mechanism of A1and D-A1on ACR-induced damage in IPEC-J2 cells. This is because A1underwent isomerization to mainly produce several procyanidin A-type dimers in simulated gastrointestinal digestion, and there were no obvious changes in the active functional groups (those bearing a catechol structure in the B ring)[18]. It can also be inferred that A1is an excellent detoxicant against ACR, because it can resist the adverse effect of gastrointestinal digestion. Considering the different absorption and metabolism of PC between the cell and animal models, it is necessary to confirm the inhibitory effect on ACRinduced damage in intestinal epithelial cells and explore its molecular mechanismin vivo.
5. Conclusion
Our findings demonstrate the protective effects of A1and D-A1against ACR-induced intestinal damage through inhibition of oxidative stress. These protective effects are largely dependent on the activation of the Nrf2 pathway via disrupting Nrf2-Keap1 interaction to facilitate Nrf2 nuclear translocation and then increase the expression of antioxidant proteins. Importantly, A1under gastrointestinal digestion possesses an excellent detoxification effect against ACR. Therefore, A1may be a promising dietary supplement for mitigating ACR-induced intestinal damage.
Conflict of interests
The authors declare that there is no conflict of interest.
Acknowledgment
This work was supported by the project from National Natural Science Foundation of China (31671962) and Fundamental Research Funds for the Central Universities (2662019PY034). The authors are grateful to Prof. Zuoxiong Liu for his valuable advice on the article.

- 食品科学与人类健康(英文)的其它文章
- Betalains protect various body organs through antioxidant and anti-inf lammatory pathways
- Effects of Maillard reaction and its product AGEs on aging and age-related diseases
- Characterization of physicochemical and immunogenic properties of allergenic proteins altered by food processing: a review
- Polyphenol components in black chokeberry (Aronia melanocarpa)as clinically proven diseases control factors—an overview
- Food-derived protein hydrolysates and peptides: anxiolytic and antidepressant activities, characteristics, and mechanisms
- Recent advances in the study of epitopes, allergens and immunologic cross-reactivity of edible mango