
Removable Dyes—The Missing Link for In-Depth N-Glycan Analysis via Multi-Method Approaches
2024-01-22 10:32SmntCjicRenHennigVlerinGroteUdoReichlErdmnnRpp
Engineering 2023年7期
Smnt Cjic, René Hennig, Vlerin Grote, Udo Reichl,c, Erdmnn Rpp,*
a Max Planck Institute for Dynamics of Complex Technical Systems, Magdeburg 39106, Germany
b glyXera GmbH, Magdeburg 39120, Germany
c Otto-von-Guericke University, Chair of Bioprocess Engineering, Magdeburg 39106, Germany
Keywords:Glycoproteins N-glycans Reversible labeling Hydrophilic interaction liquid chromatography Capillary gel electrophoresis Mass spectrometry
ABSTRACT As the roles of glycans in health and disease continue to be unraveled, it is becoming apparent that glycans’immense complexity cannot be ignored.To fully delineate glycan structures,we developed an integrative approach combining a set of cost-effective, widespread, and easy-to-handle analytical methods.The key feature of our workflow is the exploitation of a removable fluorescent label—exemplified by 9-fluorenylmethyl chloroformate (Fmoc)—to bridge the gap between diverse glycoanalytical methods,especially multiplexed capillary gel electrophoresis with laser-induced fluorescence detection (xCGELIF) and matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOFMS).Through the detailed structural analysis of selected, dauntingly complex N-glycans from chicken ovalbumin, horse serum, and bovine transferrin, we illustrate the capabilities of the presented strategy.Moreover, this approach ‘‘visualizes” N-glycans that have been difficult to identify thus far—such as the sulfated glycans on human immunoglobulin A—including minute changes in glycan structures, potentially providing useful new targets for biomarker discovery.
1.Introduction
When Karl Landsteiner mixed serum and red blood cells from six of his colleagues in 1900 [1], he observed an agglutinating effect, which was attributed many years later to an essential feature of glycoconjugates: Small changes in glycan structures can have a huge impact.In the case of Landsteiner’s discovery, the presence or absence of only one N-acetyl group on the penultimate monosaccharide of erythrocyte glycoconjugate chains (a small change) created an important difference between A and B cells,leading to blood group incompatibility (a huge impact) [2].As our knowledge of glycan structures and functions increases, we find more examples confirming that subtle differences in glycans translate to important (patho)physiological effects [3].Although the glycans on cancer cells often differ from those on normal cells by only minor alterations in structure (e.g., the position and linkage of fucose (Fuc), galactose (Gal), sialic acid (SA), or the position of antennae),they play fundamental roles in cancer formation and progression.For example, an increase in the β1-6-Nacetylglucosamine (GlcNAc) branching (but not β1-4) of triantennary N-glycans is one of the most common N-glycan structural changes found in cancer and is correlated with increased metastatic potential[4–6].Therefore,detailed N-glycan structural characterization that goes beyond compositional analysis is essential for a deeper understanding of glycan roles in biological processes,as well as for the pursuit of specific glyco-biomarkers.
A plethora of capillary electrophoresis (CE)-, liquid chromatography (LC)-, and mass spectrometry (MS)-based methods have been developed for the analysis of N-glycans.These methods differ not only in terms of their required expertise and running and maintenance costs but also in the type and amount of structural information they provide.MS detection gives direct mass information (glycan composition) but typically cannot resolve glycan structural isomers without extensive fragmentation experiments,which are sometimes considered too sophisticated [7,8].In contrast, separation-based methods coupled to optical detection have advantages in terms of simplicity and the resolution of linkage and positional isomers;however,their use in structural identification—especially of closely related, potentially overlapping N-glycans—is limited to the use of glycan standards and to enzymatic degradations [9,10].As a result, none of the existing methods alone can provide all structural information in a comprehensive manner.Therefore, detailed N-glycan structural characterization remains a demanding analytical challenge.
While current research efforts often focus on finding a single‘‘winning” glycomics method, we believe that only bringing together the advantages of various glycoanalytical methods can untangle the full complexity of glycan structures.One of the major impediments is the widespread irreversible fluorescent labeling of glycans by reductive amination.Although certain N-glycan labels can improve detection by one method, they can be detrimental to other methods[11,12].Thus,we sought to combine‘‘uncombinable”methods by exploiting cleavable fluorescent dyes.As a proof of concept, we repurposed the 9-fluorenylmethyl chloroformate(Fmoc) label, which is widely adopted as an amino-protecting group in peptide synthesis [13] and for the fabrication of glycan libraries [14] and arrays [15].This paper outlines the concept of this new analytical approach and discusses potential bottlenecks and their solutions.In addition, the benefits of this removabledye strategy are demonstrated for N-glycans with challenging structures (hybrid- and multiply-sialylated complex-type Nglycans) and modifications (O-acetylation and sulfation).Finally,even difficult-to-characterize yet biologically and clinically relevant N-glycans can be unraveled by linking fractionation for the reduction of a glycan sample’s complexity (provided by hydrophilic interaction high-performance liquid chromatography with fluorescence detection (HILIC-HPLC-FLD)) with high-throughput and high-resolution separation (provided by multiplexed capillary gel electrophoresis with laser-induced fluorescence detection (xCGELIF)) and the fast determination of glycan composition (provided by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF-MS)).
2.Methods
2.1.Materials
All utilized materials are listed in Appendix A.Aqueous solutions are indicated with the subscript ‘‘aq.”
2.2.General approach
Glycans are poorly detectable by spectroscopic methods.To make them detectable by fluorescence detectors, glycans are first derivatized with Fmoc.The fractionation of Fmoc-labeled glycan mixtures via HILIC-HPLC-FLD reduces the complexity for further analysis by xCGE-LIF, MALDI-TOF-MS, or any other method.Due to the use of the cleavable dye Fmoc, fractionated glycans can be analyzed using appropriate method, regardless of whether they are Fmoc-labeled,unlabeled,or labeled with method-specific dyes,such as 8-aminopyrene-1,3,6-trisulfonic acid (APTS) for xCGE-LIF,2-aminobenzamide (2-AB) for HILIC-HPLC-FLD analysis, or any other label(a comprehensive overview of further labeling possibilities, including reductive amination and glycosylamine-based labeling,has been provided in a recent review[16]).Thus,aliquots of fractions can be analyzed with complementary methods in parallel to obtain a full spectrum of structural information.All steps have been carefully optimized to increase the amount of released glycosylamines, labeled glycans, and recovered fractions.The final optimal conditions are given below.The experimental workflow is schematically presented in Fig.1 (please see Refs.[17–19] for the drawing of glycan structures).
2.3.N-glycan release from glycoproteins
N-glycans were enzymatically released from glycoproteins using peptide N-glycosidase F (PNGase F).As Fmoc labeling requires glycosylamines, and reductive amination-based labeling(e.g., APTS and 2-AB) requires glycans with a free reducing end,two N-glycan release strategies were followed.PNGase F release for glycan labeling via reductive amination and successive xCGELIF analysis were performed as previously reported [20,21].
To allow Fmoc labeling,the reaction conditions were optimized to obtain the highest yield of glycosylamines while restricting the hydrolysis to reducing oligosaccharides to a minimum.Of the glycoprotein-containing sample, 10 μL was reconstituted in 50 mmol∙L-1sodium phosphate buffer solution at pH 8.5, mixed with 20 μL of 2% sodium dodecyl sulfate (SDS) in 50 mmol∙L-1phosphate buffer at pH 8.5 (w/v) and incubated for 10 min at 60 °C.Subsequently, the remaining SDS was neutralized by the addition of IGEPAL CA-630 (IGEPAL), and N-glycans were released from the denatured and linearized glycoproteins by the addition of PNGase F.For this purpose, a mixture of 10 μL of 8% IGEPAL in 50 mmol∙L-1phosphate buffer at pH 8.5 (v/v), 9.5 μL of 50 mmol∙L-1phosphate buffer at pH 8.5, and 0.5 μL of PNGase F(1 unit (U)∙μL-1in 50 mmol∙L-1sodium phosphate buffer at pH 8.5) was prepared and added to each sample.The samples were incubated at 37 °C for 5 min to release the N-glycans as glycosylamines.
2.4.Fmoc labeling of released N-glycans
To make the glycans detectable by fluorescence detectors for the ensuing fractionation, enzymatically released glycosylamines were chemically modified with the fluorescent dye Fmoc.Labeling of the released N-glycans was performed for 1 h at 37°C by mixing 50 μL of N-glycan solution with 150 μL of water and 100 μL of 20 mmol∙L-1Fmoc in acetonitrile (ACN).Each sample consisting of Fmoc-labeled N-glycans was evaporated to dryness by a centrifugal evaporator.Next, 100 μL of 80% ACNaq(80% ACN:20%water;v/v)was added to the dried samples prior to further purification by means of Bio-Gel P10 (BioGel) hydrophilic interaction solid-phase extraction (HILIC-SPE).
2.5.Purification of Fmoc-labeled N-glycans
Post-derivatization sample clean-up was performed using BioGel HILIC-SPE to remove the free Fmoc,deglycosylated protein,salts, and other impurities.A volume of 200 μL of a 100 mg∙mL-1BioGel suspension in water/ethanol/ACN (70%:20%:10%; v/v/v)was added to each well of a 96-well hydrophilic membrane filter plate.Solvents were removed by vacuum using a vacuum manifold(Merck Millipore,Germany).All wells were prewashed three times with 200 μL of water, followed by equilibration three times with 200 μL of 80% ACNaq(v/v).After the Fmoc labeling procedure, the samples (in 80% ACNaq) were loaded into the wells containing BioGel suspension and shaken at 450 revolutions per minute(rpm) for 5 min to improve glycan binding.For purification, the wells were subsequently washed five times using 200 μL of 80%ACNaq.All washing steps were performed by the addition of solutions, incubation for 2 min, and the removal of solvent via a vacuum.For elution, 100 and 200 μL of water were applied to each well once and twice, respectively, followed by 5 min incubation at 450 rpm (after each addition of water).The eluates were collected in a 96-well storage plate by applying a vacuum.Combined eluates containing Fmoc-labeled N-glycans were either fractionated and/or analyzed immediately via HILIC-HPLC-FLD, or stored until fractionation/analysis at-20°C.Fmoc-labeled N-glycan samples prepared using this approach were also subjected to direct MALDI-TOF-MS analysis.If necessary, samples were concentrated through vacuum centrifugation and reconstituted in water prior to fractionation/analysis.
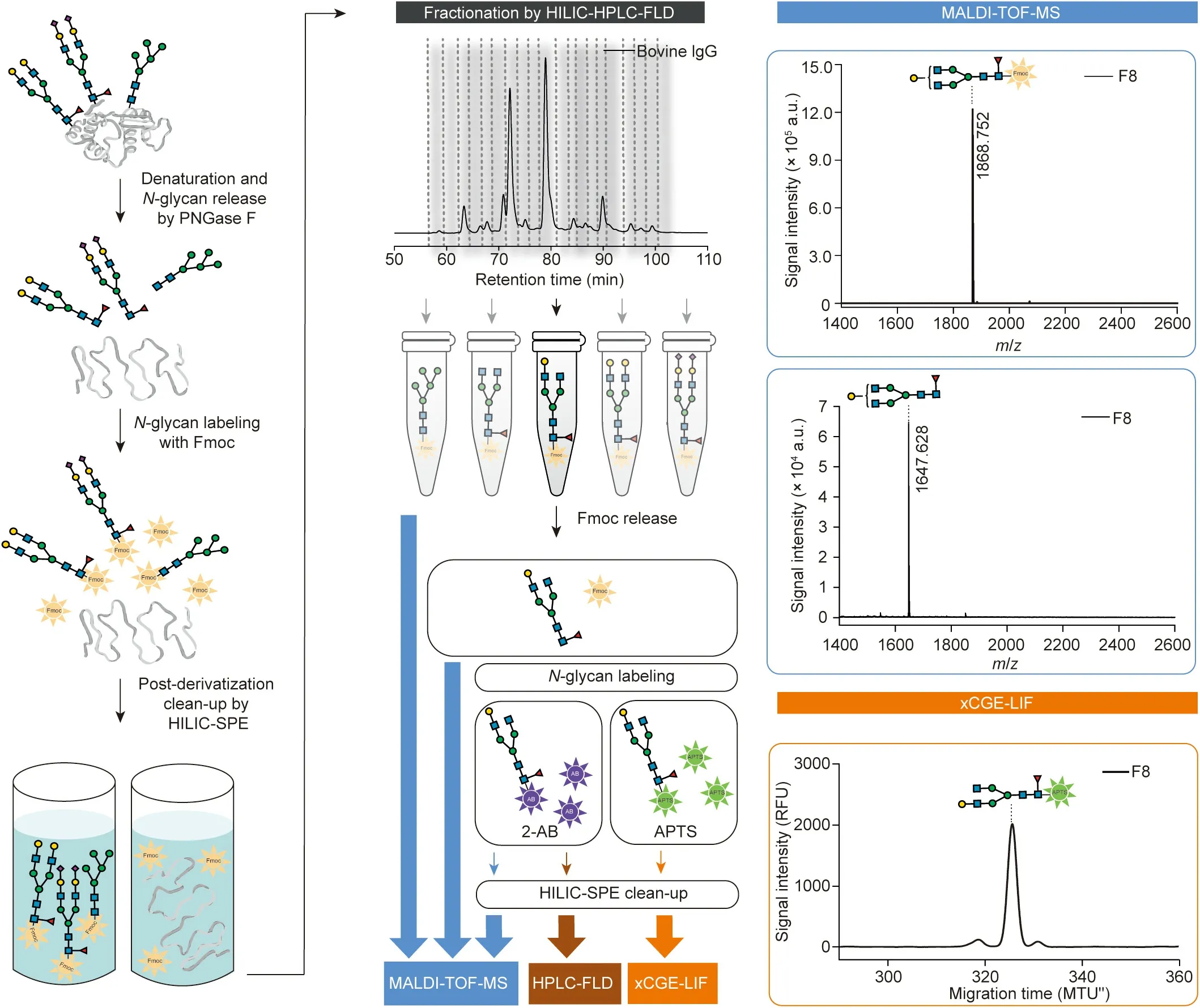
Fig.1.Analytical approach for complete structural characterization of N-glycans.Workflow involves enzymatic release of N-glycans from glycoproteins,Fmoc derivatization and subsequent purification (left panel), reduction of sample complexity by N-glycan separation using HILIC-HPLC-FLD, followed by parallel analysis of collected fractions using a combination of methods (e.g., xCGE-LIF for structural analysis and MALDI-TOF-MS for compositional analysis).Fmoc group can be released, and glycans analyzed unlabeled or after labeling with other fluorescent dyes(e.g.,APTS for xCGE-LIF or 2-AB for HILIC-HPLC-FLD and MALDI-TOF-MS)(middle panels).Example analysis of bovine immunoglobulin G (IgG) N-glycan fraction by MALDI-TOF-MS and xCGE-LIF is illustrated (right panels).N-glycan structures were drawn using the GlycanBuilder [17,18],following Symbol Nomenclature for Glycans (SNFG) guidelines [19].PNGase F: peptide N-glycosidase F; HILIC-SPE: hydrophilic interaction solid-phase extraction;a.u.: arbitrary units; m/z: mass-to-charge ratio; MTU′′: aligned migration time units; RFU: relative fluorescence units; F8: fraction 8.
2.6.Fractionation of Fmoc-labeled N-glycans via HILIC-HPLC-FLD
Fmoc-labeled and purified glycans were separated via HILICHPLC, and fractions were collected based on observed individual peaks.A volume of 90 μL of Fmoc-labeled and purified sample(diluted to 80%:20% ACN/aqueous sample; v/v) was separated on a TSKgel Amide-80 column (5.0 μm, 250.0 mm length × 4.6 mm inner diameter column; Tosoh Bioscience, Germany) at 30 °C.The setup was equipped with a guard column (10.0 mm× 4.6 mm) containing the same stationary phase and particle size as the separation column.The flow rate was set to 0.4 mL∙min-1,and a binary gradient (linear gradient; curve 5) was applied using 50 mmol∙L-1ammonium formate at pH 4.4 as the aqueous solvent A and ACN as the organic solvent B.The following gradient conditions were used: 0 min at 80% solvent B; 152 min at 42%solvent B; 155 min at 0 solvent B; 162 min at 0 solvent B;163 min at 80% solvent B; and 200 min at 80% solvent B.The samples were maintained at 4 °C before injection.Chromatographic separation and fractionation were performed on a Thermo Scientific UltiMate 3000 Rapid Separation Binary system(Dionex/Thermo Fisher Scientific, Germany) coupled to an FP-2020 Plus fluorescence detector (Jasco, Germany) with excitation and emission wavelengths of 266 and 310 nm, respectively.Instrument control was performed using Chromeleon 6.8 (Thermo Fisher Scientific) chromatography data software.Fraction collection was triggered by the fluorescence signal threshold and peak slope (with a threshold of -40 relative fluorescence units (RFU),upslope of 0.5 RFU∙s-1, and downslope of -0.5 RFU∙s-1).To reduce the quantity of ammonium formate salts, the fractions were diluted with water and dried by vacuum centrifugation.The fractions were then either immediately processed for analysis(by xCGE-LIF, MALDI-TOF-MS, and/or HILIC-ultra-highperformance liquid chromatography (UPLC)-FLD) or stored at -20 °C until analysis.
2.7.HILIC-UPLC-FLD analysis of Fmoc-labeled N-glycans
Fmoc-labeled and purified glycans were quantitatively profiled(relative quantification) by means of HILIC-UPLC-FLD.Fmoclabeled N-glycans were separated via HILIC on a Thermo Scientific UltiMate 3000 Rapid Separation Binary system (Dionex/Thermo Fisher Scientific) configured with a fluorescence detector set with excitation and emission wavelengths of 266 and 310 nm, respectively.The instrument was under the control of Chromeleon 7.2 software, SR4 (Thermo Fisher Scientific).The Fmoc-labeled N-glycans were separated on an ACQUITY UPLC BEH Glycan column (1.7 μm,150.0 mm length × 2.1 mm inner diameter column; Waters, Germany)at 40°C,with 50 mmol∙L-1ammonium formate at pH 4.4 as the aqueous solvent A and ACN as the organic solvent B.A linear gradient(curve 5)of 78.0%–55.9%of solvent B was used at a flow rate of 0.4 mL∙min-1in a 38.5 min analytical run.The ACQUITY BEH Glycan VanGuard pre-column(1.7 μm,5.0 mm×2.1 mm;Waters)was connected directly to the inlet of the ACQUITY UPLC column.An injection volume of 10–20 μL was used throughout for the samples,which were prepared in 80% ACNaq(v/v).The samples were maintained at 10°C prior to injection.Data was subsequently processed(i.e.,peak picking,integration,relative quantification,and retention time alignment)using the Java-based glycan analysis software glyXtoolLC(closed beta version;glyXera,Germany).
2.8.Release of Fmoc group from Fmoc-labeled N-glycans
To allow labeling with other dyes(e.g.,APTS labeling for xCGELIF or 2-AB for HILIC-UPLC-FLD analysis),as well as the analysis of free glycans (e.g., via MALDI-TOF-MS), it was necessary to release the Fmoc group from the glycans first.Free N-glycans were recovered by mixing 20 μL of Fmoc-labeled N-glycan-containing solution (or HILIC-HPLC fraction) with 30 μL of dimethylformamide and 20 μL of morpholine, and then incubating for 30 min at 37 °C.Before the reductive amination-based labeling of N-glycans with other fluorescent dyes (e.g., APTS or 2-AB), the samples were dried with a centrifugal evaporator.For MALDI-TOF-MS analysis,unlabeled glycans were purified using cotton HILIC-SPE and analyzed directly or after linkage-specific SA esterification (for experimental details, see Sections 2.13–2.15).
2.9.APTS labeling of free N-glycans (after release of Fmoc group)
To enable electrophoretic migration and subsequent detection,Fmoc-free glycans were fluorescently derivatized with negatively charged APTS by reductive amination.Prior to fluorescent labeling,it was necessary to convert the glycosylamine form of the released N-glycans to the reducing glycan form.For this purpose, 2 μL of 2 mol∙L-1aqueous citric acid (CAaq) was added to the dried material and, after brief centrifugation, left at room temperature for 5 min before proceeding with the next step.Labeling of the Fmoc-free N-glycans was performed for 3 h at 37°C by the addition of 2 μL of APTS Solution and 2 μL of ReduX Solution (a reductive agent), both originating from a glyXprep kit (glyXera).Alternatively,2 μL of 5 mol∙L-1CAaqwas premixed with 2 μL of APTS Solution and 2 μL of ReduX Solution, and the mixture was added directly to the dried material.To stop the labeling reaction,100 μL of Stopping Solution(from the kit)was added,and the sample was mixed carefully.
2.10.Purification of APTS-labeled N-glycans
To remove the excess APTS label, reducing agent, and other impurities, and to reduce the salt concentration, glyXbeads HILIC-SPE was employed.A sample clean-up was carried out with the glyXprep sample preparation kit (glyXera) according to the manufacturer’s instructions.In brief,the sample(in Stopping Solution) was placed in a well in a filter plate containing 200 μL of glyXbeads Slurry and incubated for 5 min at ambient temperature for binding, followed by washing and elution steps.If necessary,eluates containing purified APTS-labeled N-glycans were dried using a vacuum concentrator and dissolved in an appropriate volume of water.The samples were stored at -20 °C until analysis.
2.11.xCGE-LIF analysis of APTS-labeled N-glycans
To allow detailed structural characterization of the glycans,APTS-labeled and glyXbeads HILIC-SPE-purified glycans were analyzed by means of xCGE-LIF.The xCGE-LIF measurement of purified fluorescently labeled oligosaccharides was performed as previously reported in detail [20].For the processing of the xCGE-LIFgenerated data(i.e.,automated peak picking,integration,and relative quantification) and for the alignment of the migration time(tmig) to an internal standard (glyXalign GA2, unless otherwise noted; glyXera), glyXtoolCE software (glyXera) was used.In general, from the electropherogram, an N-glycan ‘‘fingerprint” (i.e.,an aligned electropherogram) was created: The signal intensity in RFU on the y-axis was plotted over the aligned tmigin migration time units (MTU′′) on the x-axis.Supported by the high reproducibility of the aligned tmig, N-glycan fingerprints of different samples were compared, and the annotation of N-glycan peaks was performed via tmigmatching to the glyXtoolCE N-glycan database.
2.12.Exoglycosidase treatment of APTS-labeled N-glycans
In addition to database matching, the annotation of the Nglycans (i.e., sequence, monosaccharide types, and linkages) was confirmed using comprehensive exoglycosidase digests and the subsequent reevaluation of N-glycan fingerprints by means of xCGE-LIF.The following exoglycosidase digests were performed:α2-3-sialidase (SiaS; recombinant from Streptococcus pneumoniae,expressed in Escherichia coli (E.coli)); α2-3,6-sialidase (SiaC;recombinant from Clostridium perfringens, expressed in E.coli);α2-3,6,8-sialidase (SiaA; recombinant from Arthrobacter ureafaciens, expressed in E.coli); β1-3-galactosidase (3GALase; recombinant from Xanthomonas manihotis, expressed in E.coli); β1-4-galactosidase (4GALase; recombinant from Streptococcus pneumoniae, expressed in E.coli); β1-4,6-galactosidase (46GALase; from jack bean); β1-2,3,4,6-N-acetylglucosaminidase (GlcNAcasexm;recombinant from Xanthomonas manihotis, expressed in E.coli);β1-2,3,4,6-N-acetylglucosaminidase (abbreviated as GlcNAcase or GlcNAcasesp; recombinant from Streptococcus pneumoniae,expressed in E.coli); and α1-2,3,6-mannosidase (MANase; from jack bean).The exoglycosidase digestions were carried out at 37 °C in buffers and under the conditions recommended by the suppliers of the enzymes.The samples were purified using glyXbeads HILIC-SPE prior to xCGE-LIF analysis (for details, see Section 2.10).Specific activity and possible side activity were carefully tested for each exoglycosidase enzyme used.
2.13.Ethyl esterification for the MALDI-TOF-MS analysis of Fmoclabeled N-glycans
To stabilize the SAs for subsequent MALDI-TOF-MS analysis, a linkage-specific SA esterification procedure was employed[22,23]: 1 μL of N-glycan solution was added to 20 μL of 0.25 mol∙L-11-ethyl-3-(3-dimethylaminopropyl)carbodiimide and 0.25 mol∙L-11-hydroxybenzotriazole in ethanol, and then incubated for 1 h at 37°C.Subsequently,20 μL of ACN was added,and the mixture was purified via cotton HILIC-SPE prior to MALDITOF-MS analysis, as described below.2.14.Purification of Fmoc-labeled N-glycans prior to MALDI-TOF-MS analysis
The purification of Fmoc-labeled glycans before MALDI-TOF-MS analysis was performed by means of cotton HILIC-SPE,as reported by Reiding et al.[22], with slight modifications.Of the Fmoclabeled N-glycan-containing sample, 6 μL was diluted with 34 μL of ACN for an organic content of 85% ACNaq(v/v) prior to purification.A pipette tip with a volume of 2–200 μL, packed with cotton wool,was conditioned and equilibrated by pipetting 40 μL of water three times,followed by 40 μL of 85%ACNaq(v/v)three times.The sample was loaded onto cotton wool by pipetting the reaction mixture 20 times up and down.The tip was washed three times with 40 μL of 85% ACNaqcontaining 1% trifluoroacetic acid (v/v) and three times with 40 μL of 85%ACNaq(v/v).The subsequent elution was performed in 20 μL of water.
2.15.MALDI-TOF-MS analysis of Fmoc-labeled N-glycans
The compositional information on the glycans was derived from MALDI-TOF-MS measurements.To acquire more specific structural information, selected glycans were fragmented using MALDI-TOF/TOF-MS/MS.For the MALDI-TOF-MS analysis,0.5 μL of 10 mg∙mL-1super-dihydroxybenzoic acid matrix in ACN/0.1% trifluoroacetic acid in water solution (30%:70%, v/v) containing 1 mmol∙L-1NaCl was spotted onto an MTP AnchorChip 800/384 TF MALDI target(Bruker Daltonics, Germany).Subsequently, 1 μL of the sample(incorporating Fmoc-labeled N-glycans)was applied onto the dried matrix layer.Alternatively,1 μL of the sample was applied onto an AnchorChip plate, together with 1 μL of 5 mg∙mL-12,5-dihydroxybenzoic acid in 50% ACNaq(v/v) containing 1 mmol∙L-1NaOH, and then dried under cold air.Unless otherwise stated,the N-glycan samples were interrogated in reflectron positive-ion(RP) mode on an UltraFlextreme MALDI-TOF/TOF-MS equipped with a Smartbeam-II laser and the flexControl 3.3 software Build 108(Bruker Daltonics).The instrument was calibrated using a peptide calibration standard and a dextran ladder (2 mg∙mL-1).For positive-ion mode, a 25 kV acceleration voltage was applied after a 120 ns extraction delay; for negative-ion mode, 20 kV and 100 ns were used,respectively.Ions between mass-to-charge ratio(m/z) 1000 and 5000 were recorded, and 15 000 laser shots were accumulated per spectrum.The laser power was adjusted to obtain high signal intensities, while retaining a clearly defined monoisotopic peak for all detectable oligosaccharide masses.MALDI-TOF/TOF-MS/MS analysis was performed using the‘‘LIFT”cell;fragment ions were generated via laser-induced disassociation in positiveion mode with a pulsed ion extraction of 80 ns and a precursor ion selector range of 0.45%.Data was processed with the top-hat filter and the adjacent-averaging algorithm using flexAnalysis version 3.3 Build 80 (Bruker Daltonics).
2.16.Mild acid hydrolysis of SAs from N-glycans
The chemical desialylation of APTS-labeled N-glycans was performed by incubating 10 μL of aqueous N-glycan solution with 10 μL of 4 mol∙L-1acetic acid for 2.5 h at 80 °C, as described elsewhere [24,25].Acetic acid was removed by evaporation using a vacuum concentrator.The sample was redissolved in water and subjected to further analysis.The completeness of the mild acid hydrolysis was carefully controlled using a standard glycan sample: APTS-labeled human citrate plasma N-glycans.
2.17.Alkaline hydrolysis of the O-acetyl modification of SAs
The chemical O-deacetylation of SAs on APTS-labeled N-glycans was performed by the addition of 1 μL of 1 mol∙L-1NaOH to 10 μL of aqueous N-glycan solution,followed by incubation for 30 min at 37 °C, as described elsewhere [26].The reaction was stopped by the addition of 1 μL of 1 mol∙L-1HCl followed by glyXbeads HILIC-SPE sample clean-up(for details,see Section 2.10).The completeness of the alkaline hydrolysis was carefully controlled using a standard glycan sample: APTS-labeled N-glycans derived from erythropoietin (produced in Chinese hamster ovary cells).
3.Results and discussion
3.1.Design of the removable-dye-based workflow
By labeling a glycan with a removable dye and combining the benefits of CE, LC, and MS, we are able to unravel even the most analytically challenging(and novel)glycan structures.In our previous work, we showed that xCGE-LIF delivers unprecedented separation capable of discriminating between closely related positional and linkage glycan isomers [21,27,28].However, even this method—despite being capable of resolving the smallest structural differences—has a finite peak capacity.Some of the major impediments in glycan analysis via xCGE-LIF are the identification of glycans with coincidently overlapping migration times and the full structural characterization of highly complex glycan pools (containing unknown structures).Therefore, there is still a distinct need to combine xCGE-LIF with other analytical approaches.
Combining one of the most effective structural identification and isomer-separation tools, xCGE-LIF, with a fast and simple composition-based approach,namely MALDI-TOF-MS,could result in a mutually advantageous and powerful alliance for detailed glycan analysis.APTS has been demonstrated to be an ideal label for xCGE-LIF,as it fulfills all requirements for a successful glycan analysis: a high fluorescent yield, sufficient negative charge for injection and fast separation, excitation and emission wavelengths corresponding to the instrument requirements, and the existence of a large database [10].Therefore, we first attempted to combine xCGE-LIF and MALDI-TOF-MS simply via APTS-glycans.Although some prior studies have reported the MALDI-TOF-MS detection of N-glycans labeled with APTS or similar dyes [29,30], we were unable to observe any APTS-labeled N-glycans in negative or positive mode (Figs.S1(a) and (b) in Appendix A).On the other hand,despite having a labeling efficiency greater than 95%[31],we were able to detect the remaining small amounts of unlabeled N-glycans(Fig.S1(b)).Our results here are in good agreement with some other reported observations [11,32].Contrarily, with xCGE-LIF,we were able to detect APTS-labeled N-glycans, even when the same samples that had previously been fruitlessly measured by MALDI-TOF-MS were drastically diluted (Figs.S1(c) and (d) in Appendix A).We cannot exclude the possibility that higher concentrations of the APTS-glycans would have allowed their detection by MALDI-TOF-MS; however, in a real-life scenario, it is more likely to deal with limited sample amounts than large amounts,making this approach inapplicable.Thus,the irreversible labeling of glycans with APTS impedes the combination of both approaches once labeling has been performed.
To combine the two high-throughput and easy-to-use methods of xCGE-LIF and MALDI-TOF-MS,it was necessary to reimagine the workflow for glycan analysis by introducing a reversible (i.e.,removable) label: namely, Fmoc.The concept of this removabledye-based integrative workflow is schematically depicted in Fig.1, and its principles are explained in detail in Section 2.1.In brief, Fmoc-derivatized glycans are first preparatively separated using HILIC-HPLC to reduce sample complexity, followed by the optional Fmoc removal and re-labeling of the glycans.By further combining MALDI-TOF-MS with xCGE-LIF and thereby linking glycan composition with sequence and linkage information, a more complete structural picture can be obtained.Moreover, Fmoc derivatives show greatly enhanced sensitivity in MALDI-MS and HILIC-HPLC (in comparison with conventional labels, Fig.S2 in Appendix A),which is an important aspect when the sample quantities are limited.
3.2.Investigation of reaction conditions
The applicability of Fmoc labels has been demonstrated before in the analysis of N-glycans via CE/LC-electrospray ionization(ESI)-MS methods [33,34].However, these methods did not use the full potential of Fmoc—namely, its reversibility.The idea that the Fmoc label can be removed in N-glycan analysis was explored by Kamoda et al.[35] in 2005.Since then, however, removable labeling has not found wide acceptance within the glycocommunity.Undoubtedly, combining multiple methods via a removable label is not trivial.Hence, various workflow modules needed to be carefully evaluated for their compatibility within the removable-dye approach.Compared with a previous report[35], we further optimized all the steps of the workflow and studied the long-term stability of such a reversible labeling reaction.The liquid–liquid extraction step was replaced by solid-phase extraction (SPE) purification to obtain higher signal intensities.In the next sections (Sections 3.2.1–3.2.3), we address the potential pitfalls of such a labeling approach and outline strategies to avoid possible sources of error; we then pay particular attention to showing for the first time in such detail the applicability of the removable-dye approach for the analysis of a wide range of Nglycans (Section 3.3).
3.2.1.N-glycan release from glycoproteins using PNGase F
The overall protocol for the enzymatic release and Fmoc derivatization of the oligosaccharide moiety of asparagine-linked glycans is visualized in Fig.2(a).N-glycans are released from a protein as labile N-glycosylamines (i.e., 1-amino-oligosaccharide) through the action of the enzyme PNGase F.The liberated Nglycosylamines are gradually hydrolyzed to free reducing-endglycans (i.e., reducing glycans) [36].
The greatest effort in the development of this method was to preserve the PNGase F-released glycans as N-glycosylamines,before they were further ‘‘degraded” to reducing glycans.Only Nglycosylamine-glycans can be labeled with Fmoc, while reducing glycans remain unlabeled (Fig.2(a)).An increased concentration of the glycosylamine form leads to an increased yield of Fmoclabeled glycans,making low sample amounts accessible for analysis.However, when performing PNGase F release as previously reported (by us and others) [21,35,37], both Fmoc-labeled and unlabeled glycans co-eluted during HILIC-HPLC fractionation and were detected by means of MALDI-TOF-MS in the same fractions.Although our goal was to decrease the number of glycan structures by fractionation, co-elution actually increased the sample’s complexity, dramatically complicating further analysis.To overcome these issues, our goal was to find enzymatic release conditions with minimal hydrolysis of N-glycosylamines, while keeping high PNGase F activity.For this purpose, we investigated the effects of incubation conditions (i.e., buffer pH, type, and concentration),PNGase F type and concentration,and incubation time.The PNGase F activity (i.e., the amount of released N-glycans) was indirectly determined via HILIC-UPLC (Fig.2(a), left panel), by evaluating the absolute peak area of Fmoc-labeled bovine immunoglobulin G (IgG) FA2G2 glycan, as shown in the orange and turquoise bars in Figs.2(b)–(d) (abbreviations explained in Appendix A).Moreover, the hydrolysis of the N-glycosylamines was tracked via MALDI-TOF-MS (Fig.2(a), right panel), as both forms—that is, the Fmoc-labeled and unlabeled glycans (i.e., free reducing FA2G2)—could be detected.Both areas were summed up to the total peak area,and the percentage of free reducing FA2G2 glycan is displayed(gray dots in Figs.2(b)–(d)).
We found that the reaction pH is the key parameter influencing PNGase F activity and glycosylamine stability.At pH 7.0,about 40%of all N-glycans are already hydrolyzed within a minute;thus,they are no longer available for Fmoc labeling (Fig.2(b)).However, a pH ≥8.5 stabilized the N-glycosylamines,resulting in a much later appearance of noteworthy amounts of reducing glycans (above 95% at > 180 min) and stable levels of the same (below 10%) for short reaction times(<60 min)(Fig.2(b)and Fig.S3(a)in Appendix A).To optimize the formation of N-glycosylamines and minimize the level of non-reactive hydrolysis product(i.e., reducing glycan),we also monitored the time course of the PNGase F digestion products at different pHs using high-performance anion-exchange chromatography with pulsed amperometric detection (Fig.S4 in Appendix A), which showed a similar outcome.These results are in agreement with the reported behavior of PNGase F and Nglycosylamines;that is,PNGase F has the highest activity between pH 7.5 and 9.5 [36] (with a maximum at pH 8.5 [38])—a pH range that proved to be relatively stabilizing for the N-glycosylamines.
As longer enzymatic reaction times result in the hydrolysis of the glycosylamines (Fig.2(b)), a short reaction is preferable.Since Rapid PNGase F promises deglycosylation in minutes,it seemed to offer an opportunity for more efficient labeling before significant degradation of glycosylamines could begin.Therefore, we tested a Rapid PNGase F against two conventional PNGase Fs (Fig.2(c)).However, because all enzymes reached maximum yields in a matter of minutes with a digestion buffer at pH 8.5, the use of Rapid PNGase F was not beneficial for the glycosylamine-labeling method(Fig.2(c)).In addition, no increase of the product was detected at higher enzyme concentrations(Fig.S3(b)in Appendix A).The small peak areas of Fmoc-FA2G2 obtained for buffers 2 and 3 might be due to a non-optimal pH during the enzymatic reaction (Fig.2(c),turquoise bars)—a result that confirmed the importance of pH in N-glycosylamine stability.
Because N-glycosylamines can also be generated in ammoniumcontaining buffers[39],we next investigated the effect of four different buffer types,their concentrations,and incubation times during PNGase F release (Figs.S3(c)–(e) in Appendix A).While different buffer types had minimal impact on the overall quantity(Fig.S3(c)), the degradation of N-glycosylamines to reducing glycans was slower in ammonium-containing buffer (not reaching 100%even after 240 min; Fig.S3(d)).This result is consistent with the observation that N-glycosylamines are more slowly hydrolyzed in the presence of ammonium bicarbonate [40].Moreover, it has been reported that reducing sugars can be converted back to glycosylamines in an excess of ammonium bicarbonate(saturated solutions) [41], which should increase the yield of Fmoc-labeled glycans.However, we saw a considerable decrease in peak area of Fmoc-labeled glycans (Fig.S3(e)), indicating that higher buffer concentrations decreased the labeling efficiency or even caused decomposition of the Fmoc derivative.
Finally, under optimized conditions (50 mmol∙L-1phosphate buffer at pH 8.5, 0.01 U∙μL-1PNGase F; incubation for 5 min at 37 °C), we maximized the recovery of Fmoc-labeled N-glycans to 99.5%, while keeping the amount of unlabeled, reducing glycans to a minimum (Fig.2(d)).Further protocol development included examining the extent of hydrolysis of the N-glycosylamines during Fmoc labeling(Supplementary result 1 in Appendix A).As biologically relevant glycans(thus, potential glyco-biomarkers) in particular are often present in low abundance, the conditions for APTS labeling after Fmoc release were carefully revisited and adjusted to further improve the labeling efficiency and sensitivity for xCGE-LIF analysis (Supplementary result 2 in Appendix A).
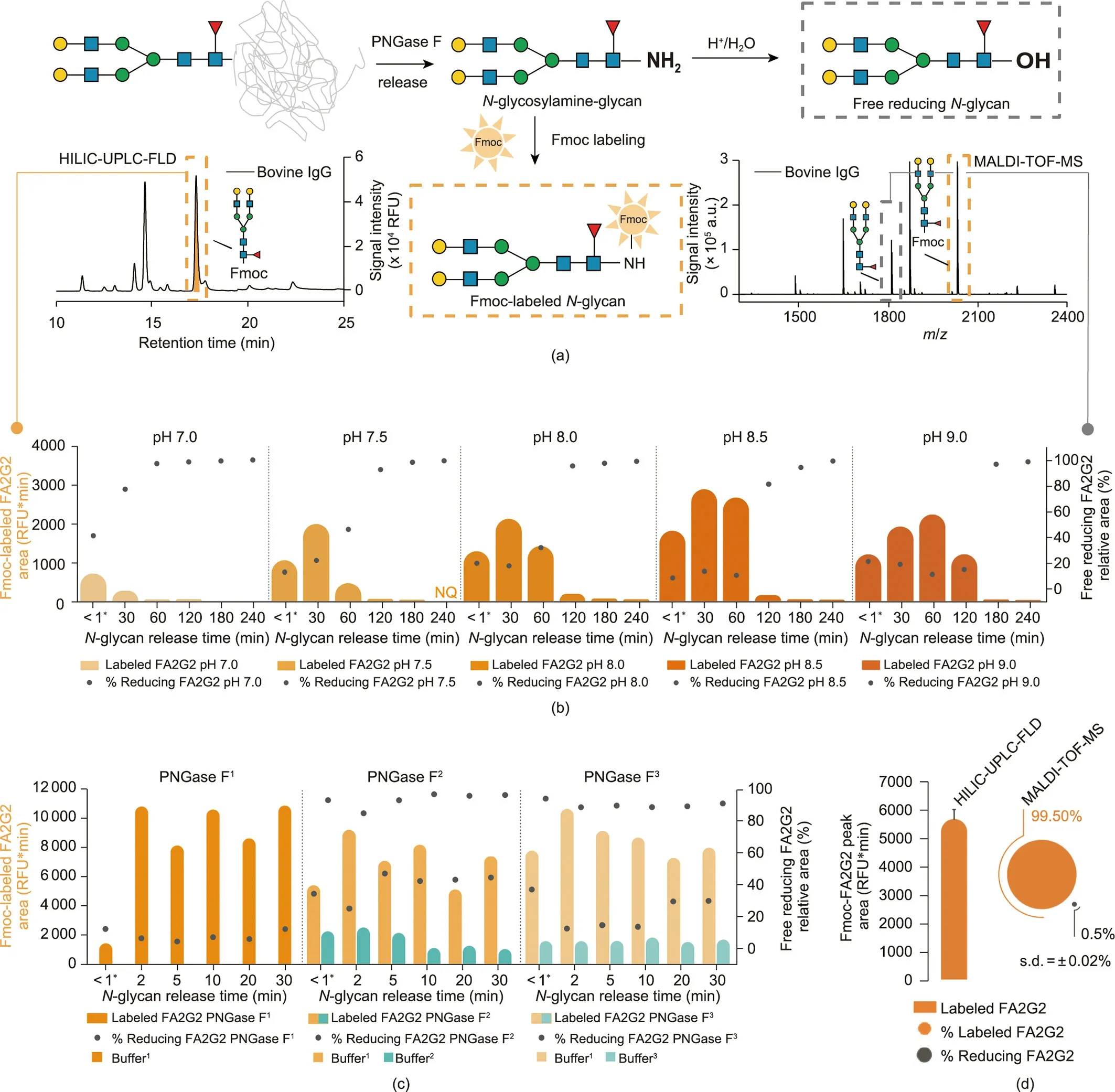
Fig.2.Optimization of N-glycan release from glycoproteins using PNGase F.(a)Reaction scheme illustrating the release of N-glycosylamine by PNGase F and its subsequent Fmoc labeling(Fmoc-labeled glycan in dashed orange box).The latter slowly hydrolyzes to a free,reducing glycan(in dashed grey box);faster under acidic conditions.HILICUPLC-FLD chromatogram:(left)amount of Fmoc-labeled N-glycans was evaluated by the peak area of the FA2G2 glycan from bovine IgG obtained by HILIC-UPLC-FLD analyses(shown as orange and turquoise bar charts in panels below).MALDI-TOF-MS spectrum (right): amount of unlabeled FA2G2 glycan remaining in the reaction mixture after labeling was evaluated by MALDI-TOF-MS;visualized as ratio of free reducing to total FA2G2(i.e.,sum of labeled and free reducing glycan(shown as grey dot charts in panels below)).(b)Effect of pH value on the number of N-glycosylamines available for Fmoc derivatization as a function of PNGase F deglycosylation time.(c)Effect of enzyme type(standard and Rapid PNGase Fs) on the number of N-glycosylamines available for Fmoc derivatization as a function of PNGase F deglycosylation time.Sigma-Aldrich P7367 PNGase F is labeled as PNGase F1, NEB P0704 as PNGase F2, and NEB P0710 Rapid PNGase F as PNGase F3.The following buffers were used for the N-glycan release:50 mmol∙L-1 sodium phosphate pH 8.5 as buffer1, buffer provided with PNGase F2 as buffer2, buffer provided with PNGase F3 as buffer3.(d) Absolute and relative yields of Fmoc-labeled and free reducing glycans,respectively,at optimized conditions for N-glycan release.Data reflect the mean and standard deviation(s.d.)of three independent experiments.*For the first sampling timepoint in(b)and(c),an aliquot of the sample was taken for Fmoc derivatization instantly after PNGase F release was started(reaction time less than a minute).For(b)–(d),PNGase F released N-glycans from bovine IgG were labeled with Fmoc and analyzed by HILIC-UPLC-FLD and MALDI-TOF-MS.The number of N-glycosylamines available for Fmoc derivatization was evaluated by HILIC-UPLC-FLD—as the absolute peak area (in RFU*min) of the Fmoc-FA2G2 peak, and by MALDITOF-MS—as the relative area of unlabeled FA2G2 (ratio of free reducing to total FA2G2; %).N-glycans are represented following the SNFG nomenclature [19].
3.2.2.Stability of Fmoc-labeled N-glycans
Apart from hydrolysis of the N-glycosylamines prior to Fmoc labeling,another origin of reducing glycans(that could complicate further analysis) could be the decomposition of already Fmoclabeled N-glycans.To investigate the stability of Fmoc-labeled Nglycans under various handling conditions, aliquots of Fmoclabeled N-glycans derived from human plasma were stored at -20 °C (comparing multiple freeze–thaw cycles), at +4 °C, and at room temperature (+21 °C), as well as in various solvents for a maximum period of two years.The absolute and relative abundances of Fmoc-labeled glycans were evaluated using HILICUPLC-FLD.The Fmoc glycan derivatives were found to be remarkably stable at different temperatures (-20, +4, and +21 °C) and in different solvents (water, organic solvent, and low-pH HILICHPLC buffer), as reflected in the constant absolute and relative peak areas of the Fmoc-labeled A2G2S2, A2G2S1, and A2G2 over time(Figs.S5(a)–(e)in Appendix A).For example,the glycan abundances did not change after storage in water at -20°C over a period of two years, even after 40 freeze–thaw cycles (Fig.S5(a)).In addition,a sample could be held in organic solvent at+4°C(conditions in the autosampler prior to fractionation) for a minimum of 40 h without affecting the glycan abundances(Fig.S5(b)).The only exception was observed when samples were stored at room temperature for more than ten days, in which case there was a decrease of sialylated glycans with a simultaneous increase of their neutral counterpart(Fig.S5(c)).However,the observed decrease of sialylated species exclusively is not caused by the decomposition of Fmoc-labeled glycans, but rather by the loss of sensitive terminal SAs,which is a known concern in glycan analysis[42].Based on these findings,the removable Fmoc label is stably linked to the glycan under a wide range of storage and handling conditions.
3.2.3.Release of the Fmoc group from N-glycans for successive relabeling
Some modern methods for glycan analysis also rely on the presence of N-glycosylamines for fluorescent labeling [43–46].However, one of the greatest benefits and strongest points of the present method—using Fmoc-labeled N-glycosylamines—arises from the application of reversible labeling.More specifically, the Fmoc group can be quantitatively removed with the weak base morpholine,quickly and easily under mild and nonselective conditions (Figs.S6(a)–(c) in Appendix A) [47].After the complexity of the glycan pool is reduced and Fmoc is removed, the glycans can be directly re-labeled with any other dye with the most favorable properties for subsequent analysis.To obtain information about a glycan structure by means of xCGE-LIF, the glycans are labeled with the APTS fluorescent dye via reductive amination.However,after the whole analytical workflow—that is, N-glycan release,Fmoc labeling and clean-up, Fmoc release, APTS labeling, and second clean-up—many additional, non-glycan-related peaks were observed when measuring via xCGE-LIF (Fig.3(a)).Non-glycanrelated peaks appeared inside the N-glycan tmigrange (region 2 in Fig.3(a)) but also outside, in the region of internal standards(regions 1 and 3 in Fig.3(a)),making both glycan analysis and tmigalignment almost impossible.Using a systematic elimination approach, we identified the source of the impurities: the morpholine (data not shown).
We further investigated whether several different brands and grades of morpholine would result in the reduction or even complete loss of the impurity peaks.Based on the electropherogram overlay (Fig.3(a)) and a comparison of the peak areas (Fig.3(b))obtained with xCGE-LIF for each commercial morpholine,we found a morpholine brand with minimal contamination peak contribution (chemical F in Figs.3(a) and (b)).An alternative approach was to purify the glycans after Fmoc release but before APTS labeling (contrary to standard procedure, which has only one purification step after APTS labeling).A liquid–liquid extraction (with diethyl ether) [35] or a SPE (BioGel and cotton HILIC-SPE) [20,22]considerably reduced the amount of impurities (Fig.3(b)).However,an additional purification step(after Fmoc release but before APTS labeling) is not practical, because it increases the hands-on time.As the amount of impurities increased with storage time for all chemicals tested, as shown for the two-year storage of chemical A in Fig.3(a)(gray trace),it is advisable to check the purity of the chemical before applying it to the sample.
3.3.Applications
Through structural analyses of selected N-glycans from chicken ovalbumin, horse serum, bovine transferrin, and human immunoglobulin A (IgA), we demonstrate the full potential of the removable-dye-based approach and its broad applicability.
3.3.1.Hybrid N-glycans
Even a minute modification of an N-glycan can have an impressive impact on biological functions and clinical relevance.For example, aberrant biantennary N-glycans bearing two GlcNAc branches on the core α1-3-mannose (Man) (and no GlcNAc on the α1-6-Man, a feature of hybrid N-glycans) were found only on glycoproteins isolated from cancer patients (e.g., choriocarcinoma and hepatocellular carcinoma) [6,48,49].Likewise, an increase in hybrid-type N-glycans and the concomitant reduction of β1-6-GlcNAc-branched structures had an effect on tumor growth and metastasis [50,51].Therefore, developing a powerful tool for Nglycan analysis that can go beyond simple composition fingerprinting and can provide a complete, detailed picture of the glycan structure is of fundamental importance.
To test whether the Fmoc-based strategy could cope with the abovementioned analytical challenges,N-glycan structures derived from chicken ovalbumin were explored in detail using the proposed method.Although ovalbumin has only a single Nglycosylation site, its glycan pool has extremely high heterogeneity,mainly containing N-glycans terminating in Man residues only(oligomannose types) and N-glycans terminating in Man and GlcNAc (hybrid types) [52].HILIC-HPLC chromatography of the Fmoc-labeled N-glycan mixture obtained from ovalbumin yielded 15 fractions (Fig.S7(a) in Appendix A), which were all subsequently analyzed by means of MALDI-TOF-MS and xCGE-LIF with exoglycosidase digestion.Fraction 8 (F8) was chosen to further demonstrate the capability of this approach to reveal detailed positional and isomeric information.
While xCGE-LIF gave rise to a single peak in F8 (Fig.S7(b) in Appendix A), MALDI-TOF-MS exposed at least two compounds with the glycan compositions hexose(Hex)4N-acetylhexosamine(HexNAc)4and Hex4HexNAc5(Fig.4(a)).Considering that Man and Gal have the same mass, and that the linkage and position of sugar residues cannot be determined by accurate mass only,these two compositions indicate the possibility of the existence of about 20 different structures.However,with the aid of the xCGE-LIF glycan database, we were able to exclude some of these structural possibilities due to tmigmismatch (Fig.4(b)).Other potentially co-migrating glycan structures with (almost) identical tmigto that of the target peak but with compositions other than those unveiled by MALDI-TOF-MS were excluded(data not shown).Consequently,the MALDI-TOF-MS results set the direction for further xCGE-LIF analysis and exoglycosidase digests.
To obtain more detailed insight into the glycan structures,exoglycosidase digests in combination with xCGE-LIF were performed.After incubation with 4GALase,the F8 peak decreased in height by approximately 25% (Fig.4(c)).This mobility change indicated that one β1-4-linked Gal was removed from the glycan by the 4GALase treatment.The degalactosylated glycan showed the same tmigas the hybrid glycan Man3-A2G0, which concurrently eliminated some other structural possibilities.The location of the Gal residue was determined with GlcNAcasexm(Fig.4(d)).After GlcNAcasexmdigestion,the F8 peak decreased in height again by approximately 25%, indicating that GlcNAc was cleaved from the same glycan as the Gal residue.Based on the tmigof the GlcNAcasexmproduct,we were able to eliminate conventional structures in which the Gal residue is located at the β1-2-linked GlcNAc attached to one of the two α-linked Man arms (Fig.4(d), green traces).Furthermore, the GlcNAcasexm-treated F8 peak (marked with an asterisk in Fig.4(d), blue trace), when subjected to a 4GALase digest,yielded a product whose tmigmismatched that of A1G0 with β1-2-linked GlcNAc (Fig.4(d), yellow traces).These findings strongly suggested that the investigated compound has a trimannosyl core with β1-4-Gal located at a GlcNAc β1-4-linked to the α1-3-Man arm and an additional unsubstituted GlcNAc β1-2-linked to the same α1-3-Man arm (Man3-A2G1; Fig.4, right panel).

Fig.3.Optimization of Fmoc release from N-glycans for successive re-labeling.(a)xCGE-LIF fingerprints(i.e.,aligned electropherograms)of APTS derivatized N-glycans from bovine IgG obtained using different sources and grades of morpholine.Peaks corresponding to the migration time alignment standard in xCGE-LIF are indicated by asterisks(*).(b) Amount of non-glycan-related peaks (i.e., impurities) obtained using different sources and grades of morpholine.The area of circles is proportional to the total peak area(in RFU*min)of the impurity signals observed in xCGE-LIF(average of three experiments).Sigma-Aldrich ReagentPlus®morpholine is designated as chemical A,Supelco analytical standard morpholine as chemical B, Millipore morpholine for synthesis as chemical C, Acros Organics morpholine as chemical D, Sigma-Aldrich ACS reagent as chemical E, and Sigma-Aldrich morpholine purified by redistillation as chemical F.For (a) and (b), Fmoc group was first released from Fmoc-labeled N-glycans and glycans were subsequently derivatized with APTS by reductive amination,purified,and analyzed by xCGE-LIF.†Additional purification was performed after Fmoc release as previously published for BioGel HILIC-SPE [20], cotton HILIC-SPE [22], and liquid–liquid extraction [35].‡Chemical stored at room temperature for two years.MTU′: MTU aligned to glyXalign GA1.
No mobility change was noticed for the second compound of the F8 peak upon 4GALase treatment, indicating the absence of Gal in this glycan (Fig.4(c)).Although this compound should contain a trimannosyl core with three GlcNAcs residues(based on the MALDI-TOF-MS), no shift by GlcNAcasexmdigest was observed(Fig.4(d)).However, GlcNAcasesphydrolyzed all three GlcNAcs,resulting in a conversion to Man4 (Fig.4(e), blue trace).Upon digestion with a mixture of 4GALase and GlcNAcasesp, the two compounds were completely converted to their Man cores of Man3 and Man4, respectively (Fig.4(e), trace with blue dotted line).The MANase digestion profile showed that the hydrolysis of the Man residues of the second investigated compound was markedly slow: After 16 h of incubation, the peak corresponding to digestion product Man3-A2BG0 increased in height by only 7%(Fig.4(f)).The results indicated that the partially digested compound was Man4-A2BG0 with a Man residue α1-3-linked to the α1-6-Man arm of the trimannosyl core,as judged by the previously reported sluggish activity of jack bean MANase toward α1-3-linked Man [53,54].In contrast, Man3-A2G1 was completely resistant to MANase digestion, which is consistent with past studies showing MANase inactivity on the Manα1-6Manα linkage of the trimannosyl core when GlcNAc extends the α1-3 arm(R-GlcNAcβ1-2Manα)[55,56].
Ovalbumin N-glycome has been scrutinized over the last 40 years,primarily on a compositional level[52,57,58].While early efforts to reveal the detailed glycan structure utilized nuclear magnetic resonance, permethylation-gas chromatography-MS, and detailed enzymatic and chemical approaches [53,54,59], these methods can be tedious and require a high degree of expertise that is only available in specialized laboratories.Here, we demonstrate the ability to pull apart numerous linkages and positional isomers through a combination of common chromatographic, electrophoretic, and mass spectrometric methods.When a 4GALase digest was performed on the total ovalbumin N-glycans, monitoring of the peak shifts was difficult(if not impossible)due to mainly minor changes in peak heights and glycan co-migration(Fig.S7(d)in Appendix A for F8-related structures).This result exemplifies how looking beyond the total N-glycome grants a more insightful picture of glycosylation, and shows that there is a practical need for combining several analytical methods.The proposed approach can unmask glycan structures that are usually unrecognized in the total N-glycome, yet are biologically relevant and thus potential biomarkers.Moreover, the results obtained for the ovalbumin hybrid glycans undoubtedly illustrate the effectiveness of the proposed approach in the analysis of various glycan structures,regardless of their complexity.The complete structural annotation of the ovalbumin N-glycans can be found in Fig.S7(e) in Appendix A.
3.3.2.O-acetylated N-glycans
SAs are recognized as an extremely diverse group of sugars[60].This diversity mostly arises from different types of O-substitutions at the 4-, 7-, 8-, and 9-hydroxyl positions of the SAs (i.e., methyl,acetyl, lactyl, sulfate, and phosphate group) [61].Changes in the O-acetylation of SAs has been observed in cancer cells [61–63],making this exquisite modification of SAs a potential diagnostic and prognostic biomarker.Unfortunately,current methods destroy(partially or completely) or miss this type of labile modification[64–66], preventing a deeper understanding of the biological roles of O-acetylated SAs.

Fig.4.Detailed structural characterization of hybrid N-glycans from chicken ovalbumin.(a) xCGE-LIF fingerprint (i.e., aligned electropherogram) and MALDI-TOF-MS spectrum of the HILIC-HPLC F8 containing ovalbumin N-glycans.(b)Aligned migration time(in MTU′′)of F8 peak was compared to those of APTS-labeled N-glycan standards with the same monosaccharide composition(hypothetical candidate structures).These standards were well characterized previously,and their migration times incorporated into the glyXtoolCE database.(c) xCGE-LIF fingerprints before and after 4GALase digest of APTS-labeled N-glycans contained in F8 and comparison with migration times of hypothetical candidate structures.(d)xCGE-LIF fingerprints before and after GlcNAcasexm digest of APTS-labeled N-glycans contained in F8 and comparison with migration times of hypothetical candidate structures.* GlcNAcasexm product (marked with asterisk in blue trace) was further subjected to 4GALase digest, giving rise to a peak delineated in the yellow trace.(e)xCGE-LIF fingerprints before and after GlcNAcasesp digest,and combined 4GALase+GlcNAcasesp digest of APTS-labeled N-glycans contained in F8.† It should be noted that GlcNAcasesp showed side-activity on Man4 with longer incubation times, breaking it down to marked (in blue trace) Man3 (confirmed by MANase digest; Fig.S7(c) in Appendix A).(f) xCGE-LIF fingerprints before and after MANase digest of APTS-labeled N-glycans contained in F8.In (b)–(d), checkboxes ⬇visualize database migration time matches(check(√)mark)and excluded structures(cross(X)mark).In(c)–(f),arrows trace the shift in the migration time of selected peaks after corresponding exoglycosidase digest:-G indicates hydrolysis of galactose;-GlcNAc indicates hydrolysis of N-acetylglucosamine;-M indicates hydrolysis of mannose residue; numbers (1x, 2x, or 3x) indicate number of hydrolyzed sugars.Right panel displays level of information obtained with each analysis step (from MALDI-TOF-MS compositional assignment to xCGE-LIF database matching and exoglycosidase sequencing).Explanation of glycan name abbreviations can be found in Appendix A.N-glycans are represented following the SNFG nomenclature [19].DB: database.
We thus wanted to evaluate the compatibility of the Fmocbased method with the analysis of a small structural modification such as SA O-acetylation.For this purpose, we chose the horse serum glycoproteome with the SAs N-acetylneuraminic acid(Neu5Ac) and N-acetyl-4-O-acetylneuraminic acid (Neu4,5Ac2) as the constituents of their N-glycan chains.Fmoc-labeled N-glycans from horse serum were submitted to chromatography on an HILIC-HPLC column, and 15 fractions were collected (Fig.S8(a) in Appendix A).Fraction 6(F6)and fraction 9(F9)were found to contain N-glycan structures with a tmigin xCGE-LIF very close to A2G2S2(6,6) and with a tmigshift of only 1.95 and 0.95 MTU′′,respectively (Fig.5(a) and Fig.S8(b) in Appendix A).A small tmigshift indicates that these two fractions contain no A2G2S2(6,6)but most probably contain closely related structures.Interestingly,the glycans in these two fractions exhibited different behavior (in terms of digestion efficiencies) upon SiaA treatment (Fig.5(b)).These observations prompted us to determine the glycan structures of F6 and F9.
F6 was first submitted to hydrolysis by SiaA,cleaving the terminal Neu5Acs,which are α2-3-,α2-6-,or α2-8-linked.After the SiaA digest, only a minor increase in monosialylated A2G2S1(6) and neutral A2G2 (due to desialylation) was observed (Fig.5(b), left).This negligible change in signal intensities was in accordance with earlier findings that O-acetylated neuraminic acids are resistant to sialidases [67,68].However, after the non-enzymatic mild acid hydrolysis of SAs(releasing all SA types,independent of their linkage and position within a glycan), the structure of interest was completely converted to the neutral component A2G2, indicating the loss of two Neu5Ac residues (Fig.5(c), left).As it was believed that the enzymatic resistance might be due to the acetylation of SA,alkaline hydrolysis of the O-acetyl group was followed by a SiaA digest of F6.After alkaline hydrolysis, SiaA cleaved the two terminal Neu5Acs and consequently generated neutral A2G2 (Fig.5(d),left).The disialylated biantennary glycan containing two O-acetyl groups was also detected via MALDI-TOF-MS as [M - 2H + 3Na]+at m/z 2594.9 (Fig.5(e)).
The same analytical approach was applied to F9.Notably, SiaA treatment mainly resulted in the hydrolysis of one SA from a disialylated glycan, considerably increasing the signal intensity of the monosialylated counterpart (Fig.5(b), right).This partial hydrolysis indicated that only one Neu5Ac was O-acetylated, and that the SiaA digest had given rise to monoacetylated A2G2S1(6).Again,mild acid hydrolysis released all Neu5Acs, which was visible by xCGE-LIF as an increase of the A2G2 peak (Fig.5(c), right).After deacetylation and the SiaA digest, the peak of interest was completely converted into the A2G2 peak,suggesting monoacetylation(Fig.5(d), right).Furthermore, a disialylated biantennary glycan containing one O-acetyl group was observed via MALDI-TOF-MS as [M - 2H + 3Na]+at m/z 2552.9 (Fig.5(e)).
To define the position of the Neu5Ac and Neu4,5Ac2residues on the two antennae, F9 was submitted to hydrolysis by SiaA, which cleaves Neu5Ac but not Neu4,5Ac2(Fig.S8(c)in Appendix A).Afterward, a 4GALase digest was performed to release the terminal Gal residue, and alkaline hydrolysis was performed to release the Oacetyl group from the terminal Neu4,5Ac2on the other arm.The appearance of two peaks in the electropherograms corresponding to A2G2S1(6)[6] and A2G2S1(6)[3] reflected the occurrence of Neu4,5Ac2on both the α1-6-Man and α1-3-Man arms of the glycan structure (Fig.S8(c)).
With the present data, we demonstrate that, in horse serum F6 and F9, the heterogeneity is linked to modifications of the SA residues(Fig.5(f)).Early studies on horse serum protein glycosylation paid close attention to different modifications of SAs [69–72].Some newer methods that have been reported for the analysis of O-acetylated SAs rely on fragmentation techniques for deeper characterization [64,65].However, all these studies (similar to the studies on ovalbumin)employ techniques that are rather costly and/or demanding in terms of expertise/equipment.To overcome these challenges, here, we established an Fmoc-based method combining two widespread pieces of equipment in molecular diagnostics laboratories: a capillary gel electrophoresis (CGE)-based DNA analyzer and MALDI-TOF-MS.Importantly, we show that,after all the steps of our workflow—that is, de-N-glycosylation,Fmoc labeling, clean-up and fractionation, Fmoc removal, APTS labeling,second clean-up,and analysis—fragile modifications such as SA O-acetylation were not lost.Moreover,the acetylated glycans were readily identified, even when minute (i.e., microliter) volumes of a complex sample mixture (horse serum) were applied(unlike some of the abovementioned methods, which required 100–1000 times greater amounts of pure glycoprotein).Although O-acetylation is only a minor modification, the related glycosylation was analyzed down to the smallest detail here (i.e., see the isomer-specific analysis of O-acetylated SAs in Fig.S8(c)).
The presented removable-dye-based approach opens up new opportunities for investigations on N-glycans from human plasma—one of the most explored biofluids in clinical glycomics.Alterations in plasma N-glycan profiles have been found to correlate with both physiological events (e.g., aging or pregnancy) [73,74]and pathophysiological events(e.g.,type II diabetes,liver cirrhosis/fibrosis, and cancer) [75–77].Those total N-glycome analyses are still a great challenge due to the complexity of the samples,the wide dynamic range of glycosylation level,and the structural heterogeneity of glycans.Notably,low-abundance glycans in plasma that could be potentially valuable disease markers may be overlooked.By applying the proposed method on horse serum, we show that our approach is applicable for the characterization of N-glycans prepared from such a complex sample; thus, it has the potential to identify biomarkers for disease onset and progression.

Fig.5.Detailed structural characterization of O-acetylated N-glycans from horse serum.(a)xCGE-LIF fingerprints(i.e.,aligned electropherograms)of the HILIC-HPLC F6 and F9 containing APTS-labeled horse serum N-glycans versus whole horse serum (zoom-in).(b) xCGE-LIF fingerprints before and after SiaA digest of APTS-labeled N-glycans contained in F6(left)and F9(right).(c)xCGE-LIF fingerprints before and after mild acetic acid hydrolysis(AA)of SAs from APTS-labeled N-glycans contained in F6(left)and F9(right).(d)xCGE-LIF fingerprints before and after mild alkaline de-O-acetylation(-Ac)of SAs and subsequent SiaA digest of APTS-labeled N-glycans contained in F6(left)and F9(right).(e)Compositional information acquired by MALDI-TOF-MS for Fmoc-labeled horse serum N-glycans identified in F6(left)and F9(right).(f)Schematic drawing of horse serum N-glycans identified in F6 (left) and F9 (right) including the overview of enzymatic and chemical action on SAs.In (b)–(d), loss of sugar residues or functional groups(i.e.,conversion of one N-glycan structure into another)due to hydrolysis(enzymatic or chemical)is indicated by arrows:-S indicates hydrolysis of SA residue;-Ac indicates hydrolysis of O-acetyl group;numbers(1x or 2x)indicate number of hydrolyzed sugars or groups.See Appendix A for glycan structure abbreviations.N-glycans are represented following the SNFG nomenclature [19].OAc on a glycan cartoon: O-acetyl group.
3.3.3.Multiply-sialylated N-glycans
Examining the full complexity of sialylated structures requires the characterization of small structural changes, such as differences in the SA linkages to the underlying glycan chain.Furthermore, it necessitates the identification of different SA types that often differ by only a subtle modification, such as a single oxygen atom (e.g., Neu5Ac vs N-glycolylneuraminic acid (Neu5Gc)).Striking changes in the levels[78],type[79],or linkage of SAs[80]have been associated with cancer invasiveness and metastasis, highlighting the importance of an SA elucidation that goes beyond scratching the surface of SA diversity.
To gain a deeper insight into the sialylated N-glycome,we combined the Fmoc-based approach with the MALDI-TOF-MS method,utilizing a selective modification of the SA carboxyl group to discriminate between α2-3-and α2-6-linked SAs[22,23].The glycans of serum transferrins from various animal species are known for unprecedented diversity in the type, number, and linkage of SAs[70,81,82].Accordingly, bovine serum transferrin (whose glycosylation is scarcely analyzed to date) was selected to showcase the variety of sialylated N-glycans and all the analytical possibilities.For that purpose, Fmoc-labeled N-glycans from bovine serum transferrin were separated by means of HILIC chromatography into eight fractions (Fig.S9(a) in Appendix A), and aliquots of fractions were further analyzed via MALDI-TOF-MS and xCGE-LIF with exoglycosidase digestion.Here,we report the results obtained for fraction 5 (F5) (Fig.S9(b) in Appendix A), which contained various multiply-sialylated N-glycans.As an example, the approach for structural determination is explained for four N-glycans (termed glycans I–IV)that were uniquely present in this fraction(Fig.6(a)).
In order to distinguish sialyl linkage isomers with identical mass in MALDI-TOF-MS, linkage-specific ethyl esterification was performed on the Fmoc-glycan fraction.The mass difference induced by the SA derivatization method allowed the discrimination of α2-3-linked (-18.011 Daltons (Da) compared with the unmodified SA) and α2-6-linked (+28.031 Da) SAs directly from the MS spectrum [22].Consequently, we obtained information not only on the type of SA(i.e.,Neu5Ac or Neu5Gc,with a mass difference of 15.999 Da) but also on the linkage types (α2-3 or α2-6)and the number of SAs(two or three)for glycans I–IV(Fig.6(b)).No glycan tmigshift was observed in xCGE-LIF after the treatment of F5 with β1-3-specific 3GALase and β1-4-specific 4GALase (Figs.S9(c)and (d) in Appendix A), showing that all Gals were occupied with terminal SAs.A SiaA digest(trimming all SAs)revealed that all four glycans contained the biantennary structure A2G2(4,4) or A2G2(3,4) as the underlying backbone (Fig.6(c)).
Aiming to completely uncover the glycan structures,we carried out additional specific exoglycosidase digests (Figs.6(d) and (e)).The α2-3-specific SiaS digest confirmed that glycan I has one terminal α2-3-linked Neu5Ac,while glycans III and IV have a terminal α2-3-linked Neu5Gc (Fig.6(d)).The presence of an additional Neu5Ac α2-6-linked to the antenna GlcNAc of glycan I was evidenced by a high-concentration (complete) SiaC digest(SiaChigh), which cleaved all terminal α2-3- and α2-6-SAs, but no branched SAs (linked to an internal, antenna GlcNAc), as shown in Fig.6(e).
In an effort to determine the location of sialyl linkages and the arrangement of the underlying glycan chain,the glycans were subjected to a sequential exoglycosidase digest.After the removal of the terminal α2-3-linked Neu5Ac from glycan I, only 3GALase—not 4GALase—acted on the exposed arm (Figs.6(f) and (g), glycan I).This led to the finding that the α2-3-Neu5Ac on glycan I was linked exclusively to β1-3-Gal.The observation that α2-6-Neu5Ac is linked to GlcNAc on the Neu5Acα2-3Galβ1-3GlcNAcβ1-R antenna was supported by a final incubation with GlcNAcase,which could not remove the GlcNAc due to the presence of branched Neu5Ac (Fig.6(f), glycan I).The identity of glycan I was further confirmed by sequential degradation with SiaChigh, GALase(3GALase or 4GALase),and GlcNAcase(Figs.S9(e)and(f)in Appendix A).In a similar way,the conclusion that glycans III and IV contain α2-3-Neu5Gc linked to β1-3-Gal and β1-4-Gal, respectively,was obtained from stepwise digestion with SiaS and GALase(3GALase or 4GALase) (Figs.6(f) and (g), glycans III and IV).The absence of branched SA on glycan III was verified by the final step of GlcNAcase treatment (Fig.S9(g) in Appendix A).A lowconcentration (partial) SiaC digest (SiaClow) removed the terminal α2-6-Neu5Ac from glycan II, while the Neu5Gcα2-6Gal linkage remained intact (Fig.6(f), glycan II).Incubation of SiaClow-treated glycan II with 4GALase gave rise to two isomeric glycans (Fig.6(g), glycan II).The exact structures of glycans I–IV are depicted in Fig.6(h).
Due to reversible labeling,a large number of analytical methods can be potentially combined with each other, with the choice depending on the research question and instrument availability(in Appendix A:Supplementary result 3 showcases a coupling with the ‘‘gold standard” method in biopharmaceutical industry, HILICHPLC).Here,MALDI-TOF-MS with a linkage-specific SA derivatization and xCGE-LIF with a multitude of exoglycosidase digestions were teamed up in order to resolve the full complexity of sialylated glycan structures on bovine serum transferrin for the first time(for the complete structural annotation, see Fig.S9(h) in Appendix A).The MALDI technique with a relatively simple spectral interpretation and straightforward SA linkage analysis provided information on the SA core structure and the linkage of SAs to the underlying sugar in a fast and efficient way.Moreover, the xCGE-LIF method with targeted exoglycosidase digests deciphered the location of the SAs as well as the exact arrangement of the underlying sugars.As a result, the presented approach based on a cleavable dye has unlocked new and exciting possibilities for exploring the complex world of sialylated glycan species.
3.3.4.Sulfated N-glycans
Antibody glycosylation has been shown to change in numerous diseases, including autoimmune diseases, infectious diseases, and cancers, correlating with disease progression and severity[83,84].These studies have mainly tackled IgG glycosylation;however, human plasma immunoglobulin A (hIgA) glycosylation has recently gained some attention as well and has been found to be associated with rheumatoid arthritis and IgA nephropathy [85].Therefore, IgA glycosylation could be exploited for diagnostic and therapeutic approaches in the future.The fact that little is stillknown about the role of the glycosylation of antibodies other than IgG in health or disease is likely a consequence of the higher complexity of their glycosylation.Here,we applied the removable-dyebased approach to expand our understanding of IgA Nglycosylation.Our xCGE-LIF analysis in combination with exoglycosidase digests confirmed previous reports[86]showing that hIgA N-glycans are of the complex biantennary type,with high levels of sialylation and galactosylation, and possible fucosylation and bisection (Fig.7(a)).Interestingly, one peak contained an Nglycan that could not be unambiguously identified.This structure(marked with an arrow in Fig.7(a)) migrated faster than the fully sialylated biantennary glycan (thus appearing earlier in the electropherogram), indicating that this glycan structure may contain additional negatively charged groups (as more negative charges=faster migrating glycan).We isolated the unknown structure by means of the HILIC-HPLC fractionation of an Fmoc-labeled hIgA-derived N-glycan pool(Fig.S10(a)in Appendix A,fraction 16;Fig.7(b),panel I)and subjected it to detailed xCGE-LIF and MALDITOF-MS analysis (Figs.7(b)–(d)).

Fig.6.Detailed structural characterization of sialylated N-glycans from bovine transferrin.Coupling the Fmoc-based method with linkage-specific SA esterification and MALDI-TOF-MS analysis.(a)xCGE-LIF fingerprint(i.e.,aligned electropherogram)of the HILIC-HPLC F5 containing APTS-labeled bovine transferrin N-glycans(Figs.S9(a)and(b) in Appendix A).(b) SA type and linkage information for Fmoc-labeled and ethyl esterified glycans I, II, III, and IV from F5 as studied by MALDI-TOF-MS.(c)–(e) Single enzyme exoglycosidase digests of APTS-labeled glycans I–IV as analyzed by xCGE-LIF before and after: (c) SiaA, (d) SiaS, (e) high-concentration (complete) SiaC (SiaChigh)digest.(f,g) xCGE-LIF fingerprints of sequential exoglycosidase digests of APTS-labeled glycans I–IV using following enzymes: low-concentration (partial) SiaC (SiaClow),4GALase, 3GALase, and GlcNAcase.(h) Bovine transferrin glycans I–IV identified in F5.(c)–(g) Vertical arrows (grey) depict peaks whose behavior upon exoglycosidase digestion is being studied.Horizontal arrows show the movement of the peaks after each digestion:-S indicates hydrolysis of SA residue;-S(3)indicates hydrolysis of α2-3-linked SA;-G(4)indicates hydrolysis of β1-4-linked galactose;-G(3)indicates hydrolysis of β1-3-linked galactose.Colored arrows indicate the specificity of exoglycosidases for particular terminal monosaccharides.The peak marked with an asterisk(*)represents one of the internal standards spiked into the samples for migration time alignment in xCGE-LIF.For detailed explanation of structure abbreviations,see Appendix A.Following structural features cannot be described with abbreviations:β1-3-galactose can be substituted with α2-3-SA only;α2-6-Neu5Ac is linked exclusively to GlcNAc on Galβ1-3GlcNAcβ1-R antenna.N-glycans are represented following the SNFG nomenclature[19].
Sequential exoglycosidase digests in combination with xCGELIF revealed the presence and position of an additional negatively charged modification on the glycan of interest—besides the SA carboxyl group (Fig.7(b)).Two sialidases of different specificity, as well as acid hydrolysis, were introduced in parallel to distinguish the α2-3-from α2-6-linked SAs of the glycan.The SAs on the target N-glycan were sensitive to SiaA digest(Fig.7(b), panel II)and acid treatment(data not shown),releasing both α2-6-and α2-3-linked SAs,but were resistant to SiaS(Fig.S10(b)in Appendix A),releasing α2-3-linked SAs only.Thus, the SAs on the target N-glycan were found to be exclusively α2-6-linked.Furthermore, the tmigshift after desialylation corresponded to the loss of two SAs.Interestingly, digestion with two distinct galactosidases gave divergent results.First, digestion with 4GALase resulted in the release of one Gal residue (Fig.7(b), panel II).On the other hand, digestion with 46GALase resulted in the removal of two Gal residues(Fig.7(b), panel III).While the magnitude of the migration shift correlated to a loss of one and two Gal residues, respectively, the tmigof the products indicated that the negative charge was still retained on the glycan.Subsequent GlcNAcase digestion resulted in the removal of one (in the case of 4GALase; Fig.7(b), panel II)or two underlying GlcNAc residues (in the case of 46GALase;Fig.7(b), panel III).More importantly, the GlcNAcase treatment of the (completely desialylated and degalactosylated) 46GALaseproduct cut the glycan to the common trimannosyl core structure with core fucosylation (FMan3), indicating that the N-glycan not only lost two GlcNAc residues but also lost its negative charge(i.e.,there was a shift to the higher tmigregion of the electropherogram due to the hydrolysis of the charged GlcNAc);Fig.7(b),panel III, blue trace).Consequently, the localization of the modification was narrowed down to one of the two GlcNAc residues attached to the trimannosyl core.
MALDI-TOF-MS unraveled the identity of this acidic glycan modification.First, the [M - H]-ion at m/z 2086.7, which was recorded in the reflectron negative-ion (RN) mode for the desialylated fraction, suggested the presence of a sulfate modification on the biantennary digalactosylated fucosylated glycan (FA2G2; Fig.7(c)).Tandem MS (MS/MS) then provided final evidence,confirming the presence and localization of a sulfate group on the FA2G2 glycan (Fig.7(d)).Fragment ions with a sulfate group were observed in the MS/MS spectrum, further demonstrating the sulfation of the N-glycan.In addition, certain fragments provided key information on the location of a sulfate group.For example, a fragment ion at m/z 1809.7 (arising from the loss of two terminal hexose residues;shaded structure in Fig.7(d))was clearly observed in the MS/MS spectrum of the [M - H + 2Na]+at m/z 2132.0 (boxed structure), suggesting that the sulfate group was located on the GlcNAc.As presented above, this assignment was supported by an exoglycosidase experiment in combination with xCGE-LIF.Thus, thanks to the use of the Fmoc-based approach, a sulfated glycan on hIgA—previously undetected by other glycoanalytical technologies—was characterized here in detail (Fig.7(e)).
To expose further details of the glycan structure, we searched for enzymes that strictly hydrolyze sulfate or sulfated sugar.In a previous collaborative work, we determined that sulfatedependent hexosaminidase removed the intact GlcNAc-6-SO4(but not the asulfated GlcNAc) from a terminal position on an Nglycan [87].When this highly specific enzyme was applied to the glycan of interest, which had been previously digested with both SiaA and 46GALase(i.e.,a completely desialylated and degalactosylated structure),it acted upon the sulfated-GlcNAc,resulting in the appearance of a glycan with one non-sulfated GlcNAc left on the α1-6-Man arm(FA1G0[6])[87].Accordingly, the utilization of this novel tool for glycoanalytics revealed the attachment of a sulfate group on the GlcNAc of the α1-3-Man arm only, thereby opening up a way for the deeper characterization of sulfated glycans.Although the detection and characterization of sulfated glycans represents a daunting task for all analytical methods [88], our method pinpointed the exact location of the sulfate on IgAderived N-glycans.
We think that this new finding will aid future investigations into the relationship between IgA glycosylation and disease, as the sulfation of N-glycans can significantly affect processes such as biological recognition and the clearance of the proteins from the body [89,90].Yet, the effect of sulfated N-glycans on IgA is unknown.In fact, although a wide variety of protocols and methods (e.g., radioactive isotope labeling, lectin binding arrays, MS of released glycans or glycopeptides in combination with chromatography, and LC with FLD) have been applied for studying IgA Nglycosylation in health and disease,including in large cohort studies [83,91], this is the first detailed report on the sulfation of an hIgA N-glycan.The fact that the structures of IgA N-glycans have already been explored using diverse analytical methods with this sulfation being unseen justifies the need for a new glycoanalytical tool.We believe that the Fmoc-based method—especially in combination with newly discovered sulfate-specific enzymes [87]—can provide a remarkable depth of structural information and can assist in answering some of the burning questions raised regarding the sulfation of other immunoglobulins [92–94].
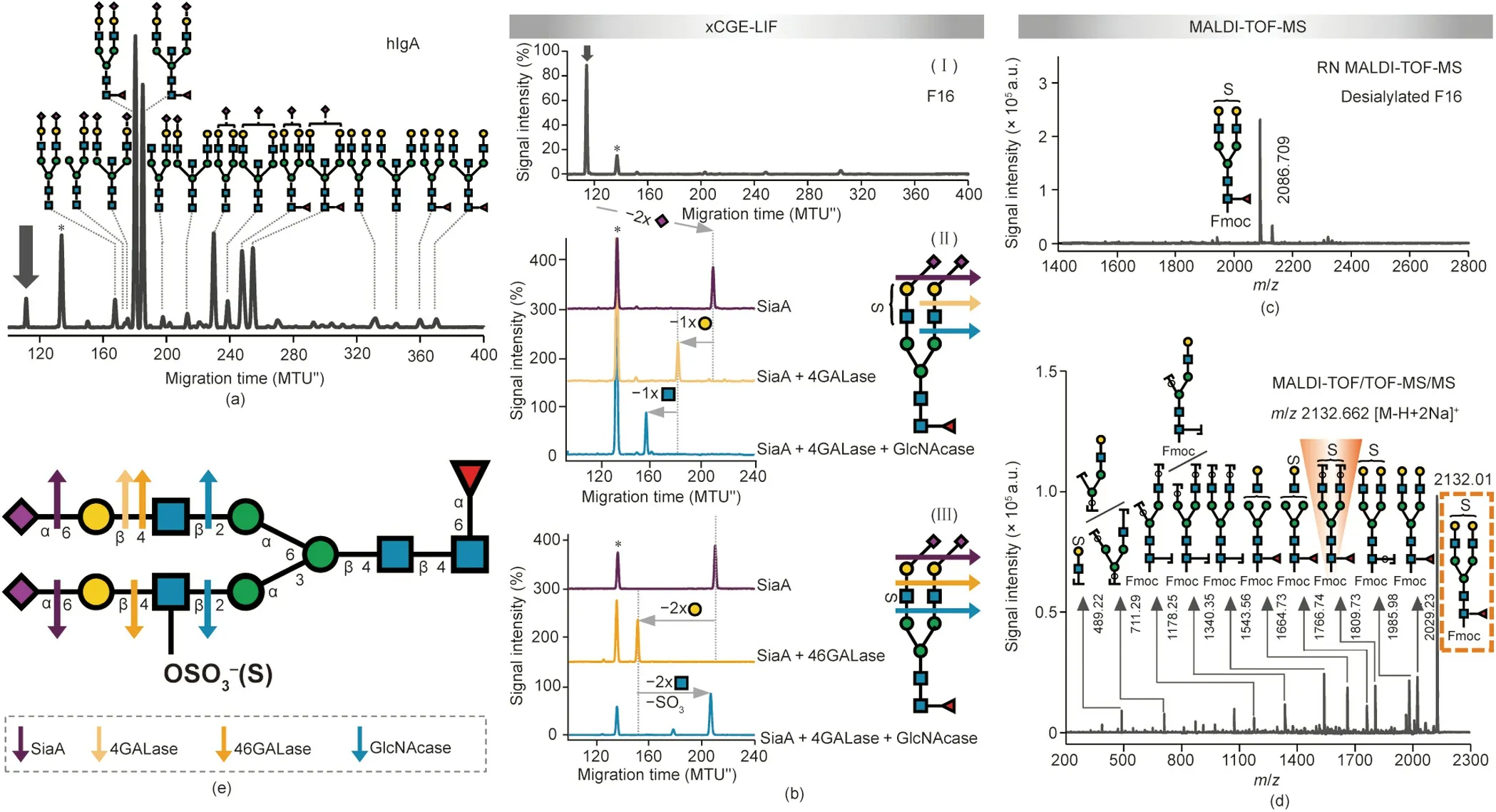
Fig.7.Detailed structural characterization of sulfated N-glycan from hIgA.(a)xCGE-LIF fingerprints(i.e.,aligned electropherograms)of the APTS-labeled hIgA N-glycans.Nglycan structures were assigned via database matching and confirmed by exoglycosidase sequencing (both in combination with xCGE-LIF).(b) Panel I shows the xCGE-LIF fingerprint of HILIC-HPLC F16 containing APTS-labeled hIgA N-glycans.Panels II and III display sequential exoglycosidase digests of APTS-labeled N-glycans contained in F16 as analyzed by xCGE-LIF.Digests were performed using following enzymes:SiaA,4GALase,46GALase,and GlcNAcase.In N-glycan fingerprints at left,the activity of enzymes is visualized by the arrows that show the migration time shift of the corresponding N-glycan.In both panels,enzyme activity is illustrated at right.(c)Reflectron negative-ion(RN) MALDI-TOF-MS spectrum of desialylated F16 when Fmoc-labeled.(d) MALDI-TOF/TOF-MS/MS spectrum of the major N-glycan in the Fmoc-labeled F16 (analyte of interest; highlighted in the dashed orange box).Key diagnostic fragment indicating localization of sulfate group is shaded orange.(e) Schematic drawing of hIgA N-glycan identified in F16.For glycan illustrations in(b)and(e),arrows indicate activity of exoglycosidases on terminal glycan residues(enzyme color legend provided in(e).The angle of the Neu5Ac-Gal bond(depicted in(b))is indicative of α2-6 linkage.The letter S on glycan cartoons in(b)–(e)marks sulfate group.Peak marked with arrow()in(a),(b)represents a so far unknown N-glycan structure (analyte of interest).Peak marked with asterisk (*) in (a) and (b) corresponds to one of the internal standards used for migration time alignment in xCGE-LIF.N-glycans are represented following the SNFG nomenclature [19].
4.Conclusions
In the last few decades, we have seen many advances in the technologies used for the structural analysis of N-glycans.However, the emphasis of these analytical workflows has mainly been on increasing the speed, throughput, and sensitivity, often at the cost of the depth and breadth of glycome analysis.To understand the biological information contained in glycosylation, it is necessary to be able to fully dissect the glycan structures first.The basic aim of this communication was to demonstrate the power of combining a high-resolution CE-based method (xCGE-LIF) with a fast and easy-to-use MS-based method(MALDI-TOF-MS)via reversible labeling to disentangle the glycan complexity.With the application of a removable label,preexisting glycan analysis technologies with advantages in simplicity, low cost, throughput, structural resolution, and sensitivity were easily integrated to unravel structural details(e.g.,positional and linkage isomers).Using chicken ovalbumin, horse serum, bovine transferrin, and hIgA as illustrative examples, we not only confirmed already known structural information that was previously accessed in such detail only through demanding analytical approaches but also unmasked previously unseen structural details.Although this paper focuses on the combination of two high-throughput methods, xCGE-LIF and MALDITOF-MS, the application of a removable dye opens the door to many new analysis routes.We believe that the approach presented here will encourage the glyco-community to consider reversible labeling as a way to combine previously ‘‘uncombinable” techniques for in-depth glycan analysis.In addition, we hope that the presented method will make glycan analysis more accessible to the broader research community and will consequently steer research toward gaining a deeper understanding of the role of glycans in physiology and disease.
Nomenclature
2-AB2-aminobenzamide
3GALaseβ1-3-galactosidase
4GALaseβ1-4-galactosidase
46GALaseβ1-4,6-galactosidase
APTS8-aminopyrene-1,3,6-trisulfonic acid
BioGelBio-Gel P10
CEcapillary electrophoresis
Fmoc9-fluorenylmethyl chloroformate
Fucfucose
Galgalactose
GlcNAcN-acetylglucosamine
GlcNAcaseorGlcNAcasespStreptococcus pneumoniae β1-2,3,4,6-Nacetylglucosaminidase
GlcNAcasexmXanthomonas manihotis β1-2,3,4,6-Nacetylglucosaminidase
(h)IgA(human) immunoglobulin A
HILIC-HPLC-FLDhydrophilic interaction high-performance liquid chromatography with fluorescence detection
IgGimmunoglobulin G
LCliquid chromatography
Manmannose
MANaseα1-2,3,6-mannosidase
MALDI-TOF-MSmatrix-assisted laser desorption/ionization timeof-flight mass spectrometry
MSmass spectrometry
MTU′′aligned migration time units
m/z mass-to-charge ratio
Neu5AcN-acetylneuraminic acid
Neu4,5Ac2N-acetyl-4-O-acetylneuraminic acid
Neu5GcN-glycolylneuraminic acid
PNGase Fpeptide N-glycosidase F
RFUrelative fluorescence units
RNreflectron negative-ion mode
RPreflectron positive-ion mode
SAsialic acid
SiaAα2-3,6,8-sialidase
SiaCα2-3,6-sialidase
SiaSα2-3-sialidase
SPEsolid-phase extraction
tmigmigration time
UPLCultra-high-performance liquid chromatography
xCGE-LIFmultiplexed capillary gel electrophoresis with laserinduced fluorescence detection
Acknowledgments
We would like to thank Silvana Fischer and Lisa Fichtmüller for technical support with the optimization experiments and Alex Behne for support with the glyXtoolCE (commercially available)and glyXtoolLC (closed beta version) software.We also thank Léa Chuzel and Marcus Hoffmann for helpful discussions and critical reading of the manuscript.We acknowledge support from the German Federal Ministry of Education and Research(BMBF)under the project ‘‘Die Golgi Glykan Fabrik 2.0” (031A557C for Samanta Cajic and Erdmann Rapp), the European Commission (EC) under the project ‘‘HighGlycan” (278535 for René Hennig and Erdmann Rapp), and the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under the project ‘‘The concert of dolicholbased glycosylation: from molecules to disease models”(FOR2509 for Valerian Grote and Erdmann Rapp).We are grateful to the glyXera GmbH for providing the software tools glyXtoolCE(commercial software) and glyXtoolLC (closed beta version) for data analysis.
Authors’ contributions
Samanta Cajic designed, planned, and performed most of the experiments, analyzed and interpreted data, and wrote the manuscript.Samanta Cajic and René Hennig prepared figures.Valerian Grote performed the experiments on horse serum and hIgA Nglycans, as well as their data analysis and interpretation.René Hennig, Valerian Grote, and Erdmann Rapp contributed to data interpretation.René Hennig proposed the project and provided expertise and feedback.Erdmann Rapp supervised the project.All authors critically revised and edited the manuscript, and confirmed the final version of the manuscript.
Compliance with ethics guidelines
The authors declare the following financial interests/personal relationships which may be considered as potential competing interests: Erdmann Rapp is co-affiliated and Samanta Cajic and René Hennig are employees of glyXera GmbH.glyXera provides high-performance glycoanalytical products and services and holds several patents for xCGE-LIF and MALDI-based glycoanalysis.
Appendix A.Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.eng.2023.02.016.

- Engineering的其它文章
- Profound Diversity of the N-Glycome from Microdissected Regions of Colorectal Cancer, Stroma, and Normal Colon Mucosa
- Twelve Years of Genome-Wide Association Studies of Human Protein N-Glycosylation
- Most Efficient Reaction Bolsters Prospects for Low-Carbon Ammonia
- Periodic Changes in the N-Glycosylation of Immunoglobulin G During the Menstrual Cycle
- Serum N-Glycan Markers for Diagnosing Significant Liver Fibrosis and Cirrhosis in Chronic Hepatitis B Patients with Normal Alanine Aminotransferase Levels
- A Chinese–French Study on Nuclear Energy and the Environment