
两个多酸基镧系(Eu、Tb)双膦酸酯配合物的合成、物化性质和太赫兹时域光谱
2024-01-20 03:55侯川兵李樱雨迟婧怡王志强韩洪亮李杏茹金琼花
无机化学学报 2024年1期
侯川兵 李樱雨 迟婧怡 丁 延 王志强 韩洪亮 李杏茹 金琼花*,,3,4,5
(1首都师范大学资源环境与旅游学院,北京 100048)
(2首都师范大学化学系,北京 100048)
(3元素有机化学国家重点实验室(南开大学),天津 300071)
(4南开大学先进能源材料化学教育部重点实验室,天津 300071)
(5中科院长春应用化学研究所稀土资源利用国家重点实验室,长春 130022)
0 引 言
稀土元素(rare earth)是17 种具有非常相似的化学和物理性质的元素的统称,包括镧系元素、钪和钇[1]。镧系金属功能配合物作为一类重要的稀土材料[2],在发光器件[3]、聚合催化[4]、医疗[5]等方面具有很大的研究空间和应用潜力。镧系金属阳离子具有非常强的氧亲和力,并且可以容易地与高极性含氧基团例如羧酸、膦氧有机物等相互作用,形成各种配合物和配位聚合物[6-7]。其中稀土膦氧配合物具有优异的性能和广阔的应用前景,在发光等方向上取得了进展[8-9]。同时,多金属氧酸盐(POMs)因其高化学稳定性、高负电、端氧丰富、尺寸及形状多样等特点,在发光、催化、生物医药等领域有着广泛应用潜力[10-14]。
多酸基金属功能配合物能光致发光的较少,其中大多数材料的发光不是来自金属离子内部的荧光发射,例如,2006年王恩波课题组研究的2个基于P4Mo6簇的镉配合物[15]和2007 年彭军课题组报道的2 个Wells-Dawson 型化合物[16]的荧光发射峰归属于配体到金属的跃迁(LMCT);2010 年薛刚林课题组报道的一例基于As2W18的金属-有机配合物的荧光发射峰归属于O→W 的电荷转移和金属到配体的电荷跃迁[17];2012 年马建方等报道的一例[Mo8O26]4-基金属配合物的荧光发射峰归属于配体内部的电荷跃迁[18]。此外也有学者报道了2 例多酸基的铕、铽配合物的荧光发射,两者的发光均是金属离子内部的电荷跃迁导致的[19-20]。
基于以上研究背景,根据软硬酸碱理论,我们已经做过一系列工作,使用膦氧配体、氮配体等和镧系金属阳离子配位,并使用Keggin 型多酸修饰,合成了一系列镧系金属配合物[21-25]。本工作中,我们使用亚乙基二膦酸四乙酯和EuCl3·6H2O、TbCl3·6H2O配位,配体和金属离子的物质的量之比为3∶1,以满足镧系金属离子的高配位数。同时在体系中加入Keggin 型多酸H3PW12O40·nH2O。晶体结构分析表明,多酸阴离子不参与配位,仅作为抗衡阴离子存在,起到模板作用。乙腈和水的混合溶剂更有利于形成高质量、高产率的晶体。本文所研究的配合物[Eu(L)4]PW12O40·2CH3CN (1) 和[Tb(L)3(H2O)]PW12O40(2)(L=亚乙基二膦酸四乙酯)则分别显示了典型的铕配合物红光发射和铽配合物绿光发射,是金属离子内部的电荷跃迁导致的,这与我们之前的工作一致[26]。
此外,我们测定了配合物1 和2 的太赫兹时域光谱(THz-TDS),该技术已被证明可以检测自由电子运动、分子旋转模式、晶格振动和偶极跃迁,从而表征分子间相互作用的方式[27]。尽管太赫兹时域光谱具有许多独特的性能,并在过去十年中被广泛应用于研究领域[28-29],但它很少用于多酸基有机-无机配合物。在之前的工作中我们已经合成了很多镧系元素配合物,并通过太赫兹光谱对其表征[26]。太赫兹光谱的应用为配合物1 和2 的结构和发光的分析提供了更多有用的信息。
1 实验部分
1.1 试剂与仪器
实验所需的试剂如表S1(Supporting information)所示。
使用Elementar Vario MICRO CUBE 元素分析仪进行元素分析(C,H 和N)。使用烘干后的KBr 压片,在Bruker EQUINOX 55 傅里叶变换红外光谱仪上进行红外光谱(FTIR)分析,波数范围为4 000~400 cm-1。使用Bruker D8 Advance 粉末衍射仪收集粉末X 射线衍射(PXRD)数据,以CuKα为射线源(λ=0.154 18 nm,U=40 kV,I=40 mA),扫描范围2θ=5°~50°,扫描速度为5(°)·min-1。使用岛津DTG-60热重分析仪在N2氛围中进行热重分析(TGA),分析范围为30~800 ℃,升温速度为10 ℃·min-1。使用日立F-4500 发光分光光度计记录固态荧光光谱。使用英国爱丁堡公司生产的FLS100/FS5 稳态瞬态荧光光谱仪进行光致发光量子产率的测定。以BaSO4为参比,使用普析TU-1901 进行配合物的紫外可见漫反射光谱(diffuse reflectance UV-Vis spectra)分析。使用聚乙烯将样品压片,在氮气氛围下通过太赫兹时域装置测得其光谱,测定范围为0.2~2.8 THz(6.6~92.4 cm-1)。
1.2 配合物1~2的合成
依次将0.40 mmol LnCl3·6H2O(Ln=Eu、Tb)、0.40 mmol H3PW12O40·nH2O 和1.2 mmol 配体L 加入到100 mL 圆底烧瓶,用30 mL 乙腈和30 mL 去离子水溶解,70 ℃水浴加热3 h,同时进行磁力搅拌。反应结束后,将溶液过滤到100 mL 的锥形瓶中,用保鲜膜封口,在室温下缓慢蒸发,2 周后得到大量晶体,配合物1和2均是无色透明的块状晶体。
配合物1:产率(以L 计)为75.5%。元素分析(C44H102EuN2O64P9W12)理论值(%):C,12.05;H,2.35;N,0.64。实验值(%):C,12.32;H,2.19;N,0.69。红外光谱(KBr 压片,cm-1):3 437(m),2 986(m),2 932(w),1 630(w),1 478(w),1 444(w),1 395(w),1 304(w),1 228(s),1 164(m),1 081(s),1 035(s),979(s),897(s),817(s),701(w),596(w),523(m)。
配合物2:产率(以L 计)为57.4%。元素分析(C30H74O59P7TbW12)理论值(%):C,9.10;H,1.88。实验测量值(%):C,9.03;H,1.84。红外光谱(KBr 压片,cm-1):3 491(m),2 986(m),2 928(w),1 639(w),1 477(w),1 444(m),1 411(m),1 371(w),1 303(m),1 208(s),1 162(s),1 081(s),1 034(s),979(s),896(s),820(s),596(m),521(s)。
1.3 单晶结构分析
298 K 的条件下,在单晶X 射线衍射仪(Bruker SMART CCD)上测得配合物1~2的晶体数据,以石墨单色器MoKα(λ=0.071 073 nm)收集所有独立衍射点,经BRUKER SAINT 做数据还原,随后使用SHELXS 97 和SHELXL-97 程序采用直接法进行结构的分析,采用全矩阵最小二乘法对所有非氢原子的热参数进行修正。分子空间堆积结构中的弱作用力及孔道直径采用Platon 程序计算获得[30-31]。配合物1~2 的晶体学数据及结构精修数据如表1 所示,配合物1~2的选定键长和键角如表2所示。
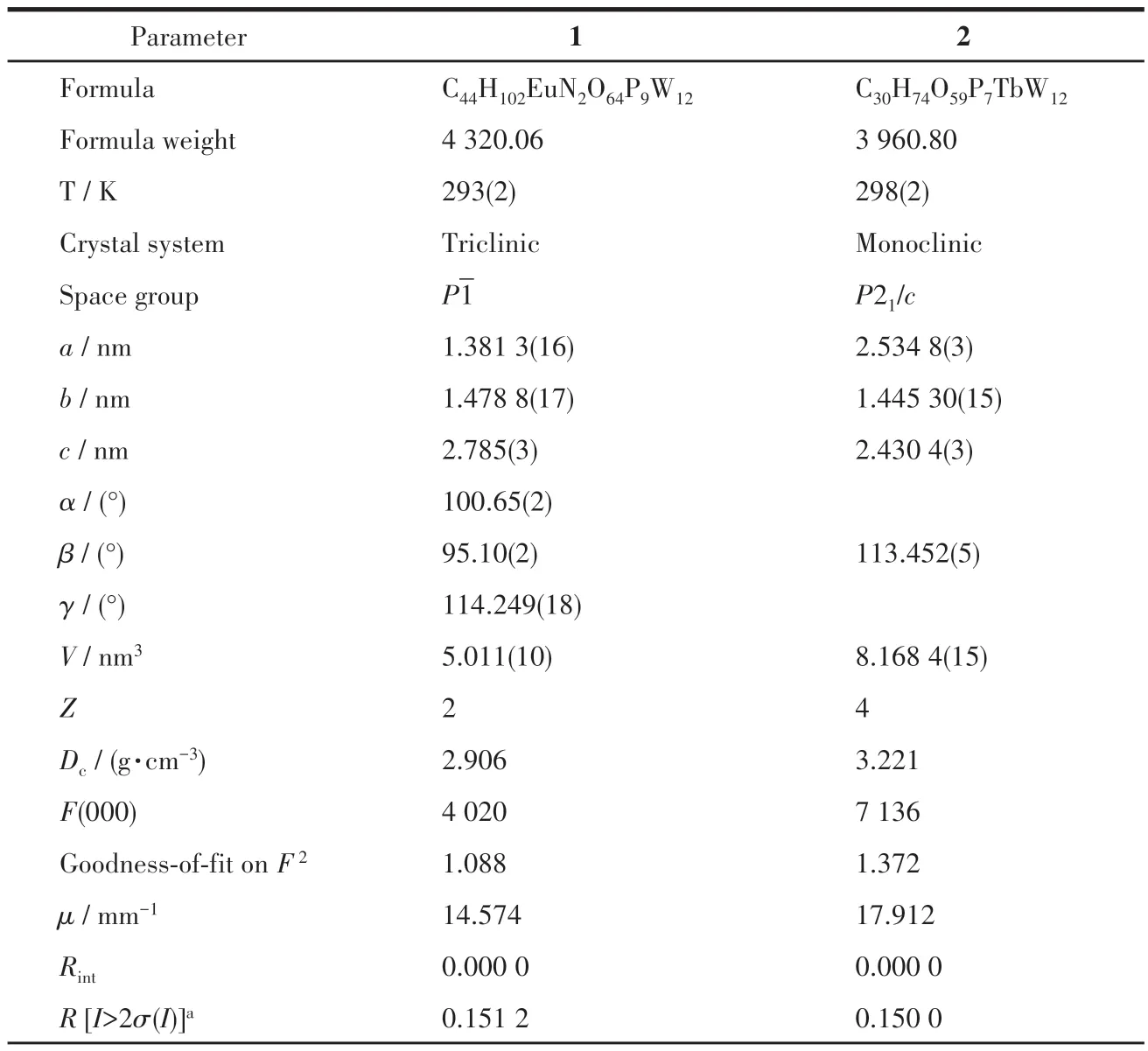
表1 配合物1~2的晶体学数据Table 1 Crystallographic data of complexes 1-2

表2 配合物1~2的选定键长(nm)和键角(°)Table 2 Selected bond length(nm)and bond angle(°)for complexes 1-2
CCDC:2321285,1;2321286,2。
2 结果与讨论
2.1 晶体结构
单晶X 射线衍射结果表明,配合物1 属于三斜晶系,P空间群。每一个不对称单元中包含了1 个[EuL4]3+阳离子,1 个未配位的[PW12O40]3-阴离子和2个CH3CN 分子(图1)。L 配体通过2个膦酸酯基团上的氧原子与中心金属Eu3+形成双齿螯合配位,Eu3+与来自4 个配体上的8 个O 原子形成一个扭曲的四方反棱柱结构(图2)。PW12O403-仅作为阴离子起平衡电荷的作用,没有参与配位;乙腈分子被固定在晶格中,同样不参与配位。Eu3+与L配体形成的Eu—O键长范围在0.231~0.245 nm 之间,与已报道的类似化合物[Ln(L1)4](OTf)3(L1=亚甲基二膦酸四异丙酯)中Eu—O 平均键长(0.239 9 nm)相近[26],配体螯合形成的(P)O—Eu—O(P)键角在77.0°~79.4°之间。
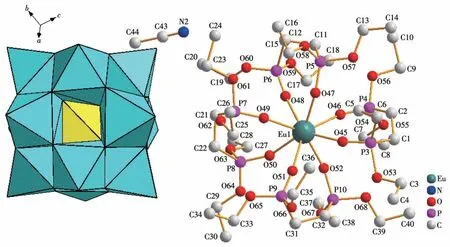
图1 配合物1的单晶结构Fig.1 Single crystal structure of complex 1
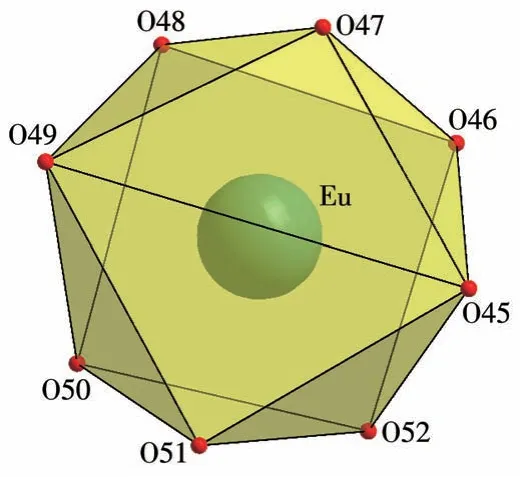
图2 配合物1的配位模式Fig.2 Coordination mode of complex 1
Platon 程序计算结果表明,配合物1 通过18 种分子间氢键弱作用,形成稳定的三维超分子结构,分子间氢键类型全部为C—H…O,其中质子供体来自L 配体或乙腈分子上的C 原子,质子受体来自多酸阴离子表面的端氧。乙腈分子通过分子间氢键被固定在多酸阴离子表面,最终稳定存在于晶格当中。每一个[PW12O40]3-阴离子和[Eu(L)4]3+作为基本结构单元,在(111)晶面上,每2 个结构单元间通过1~4对氢键相连,以ABAB 型结构交替连接,形成二维结构平面(图3),晶体的三维堆积结构在垂直于a轴和b轴的面都显示为2种结构单元交替连结;在垂直于c轴的面显示为多酸阴离子的无机层与配合物有机层交替连接(图3)。此外配合物1中还存在分子内氢键,这些分子内氢键形成于L配体中,可能影响碳链的旋转,从而对分子的堆积结构起到影响。配合物1堆积结构中的弱相互作用全部列于表S2中。
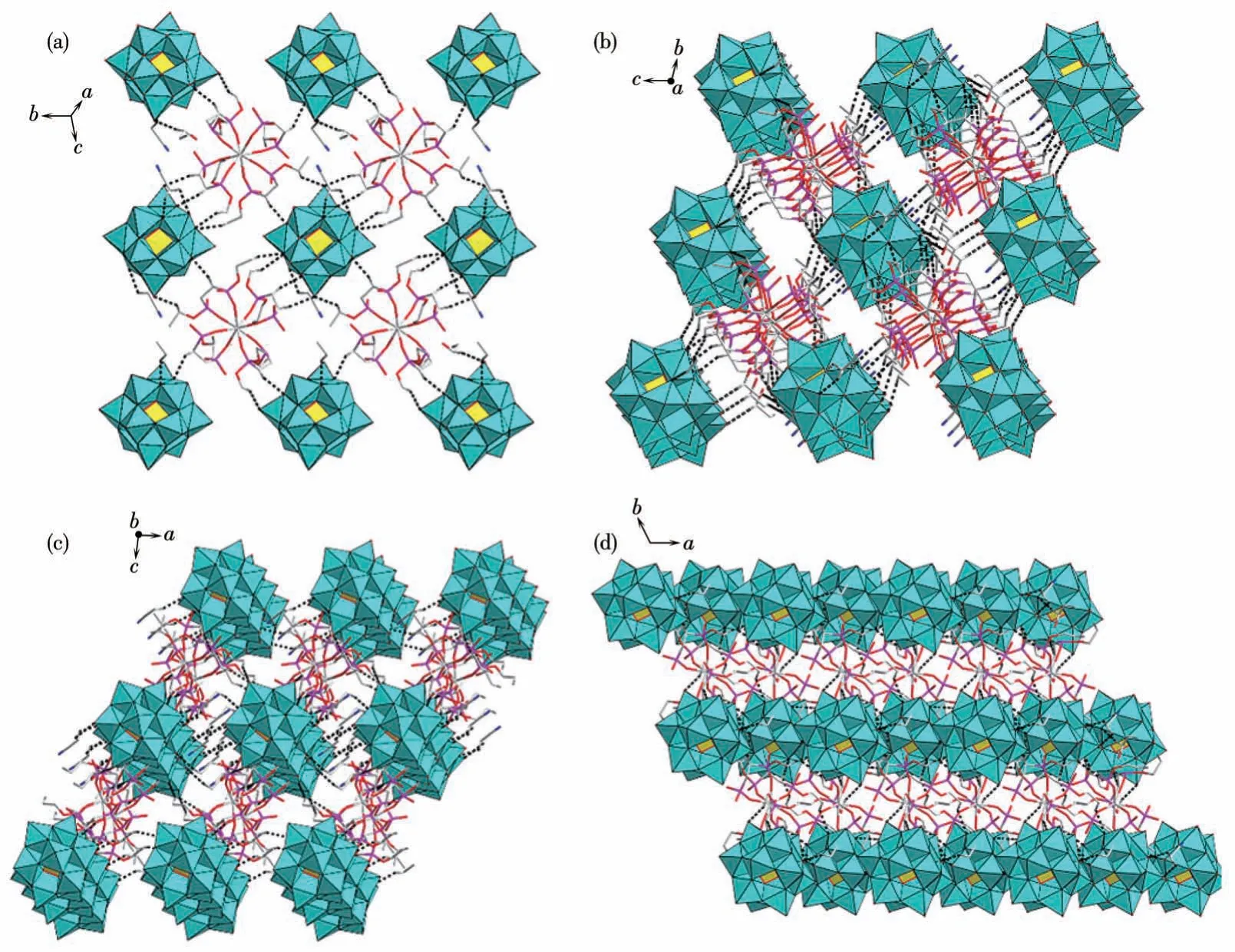
图3 配合物1中的部分氢键作用分子堆积图Fig.3 Partial hydrogen bonding and molecular packing diagram of complex 1
单晶X 射线衍射结果表明,配合物2 属于单斜晶系,P21/c空间群。每一个不对称单元中包含了1个[Tb(L)3(H2O)]3+阳离子和1 个未配位[PW12O40]3-阴离子(图4)。L 配体通过2 个膦酸酯基团上的氧原子与中心金属Tb3+形成双齿螯合配位,Tb3+与来自3 个配体上的6个O原子以及1个水分子上的O原子配位,形成一个扭曲的单帽八面体结构(图5)。[PW12O40]3-仅作为阴离子起平衡电荷的作用,没有参与配位。Tb3+与L 配体形成的Tb—O 键长范围在0.214 3~0.242 nm 之间,与已报道的相似化合物[Ln(L)2(H2O)4Cl]Cl中的Tb—O平均键长相近(0.232 4 nm)[32],配体螯合形成的(P)O—Tb—O(P)键角在76.9°~80.2°之间。
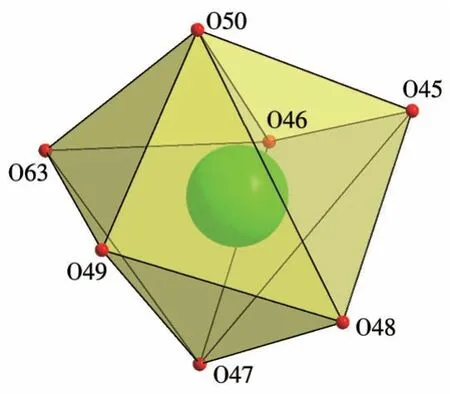
图5 配合物2的配位模式Fig.5 Coordination mode of complex 2
Platon 程序计算结果表明,配合物2 通过14 种分子间氢键弱作用,形成稳定的三维超分子结构,分子间氢键类型全部为C—H…O,其中质子供体来自L 配体的C 原子,质子受体来自多酸阴离子表面的端氧。每一个[PW12O40]3-阴离子和[Tb(L)3(H2O)]3+作为基本结构单元,在(111)晶面2 种结构单元通过ABAB型交替排列形成二维结构,配合物2中存在少数相邻结构单元间没有氢键连接的情况,但多数相邻结构单元间通过1~3 对氢键相连,与配合物1 不同,配合物2 只在垂直于b轴的面表现出2种结构单元交替连接的网状结构,在垂直于a轴和c轴的面都显示为无机层和有机层交替连接的层状结构(图6)。此外配合物2中还存在1种分子内氢键,配合物2堆积结构中的氢键弱相互作用全部列于表S3中。
对比以往的工作[25],本文将[PMo12O40]3-替换为[PW12O40]3-,使得铽配合物的选定键长变短以及铕、铽配合物的选定键角变大,对铕配合物的选定键长的大小几乎没有影响,其中铽配合物选定键角的变大,除了多酸不同的原因外,还有可能是因为配位数的不同。此外,对比配合物1和2,因为镧系收缩,铕原子比铽原子半径大,因此可以容纳更多的配体,配位数较大。
2.2 红外光谱、PXRD和TGA
在室温下使用KBr 压片测量了配合物1~2 的红外光谱(图S1),对特征吸收峰进行分析及归属。红外光谱显示,对于配体L,1 260 cm-1处为P=O 键的伸缩振动吸收峰;1 035、1 055 cm-1处为P—O—C 的振动吸收峰[33];对于磷钨酸,1 081 cm-1为P—O 键的振动吸收峰,982 cm-1处为W=O 的振动吸收峰,889 cm-1处为W—Oc—W 的振动吸收峰,797 cm-1处为W—Oe—W 的振动吸收峰,595 cm-1处为P—O 键的弯曲振动吸收峰[34]。
配合物1 主要的特征吸收峰:1 228、595、1 035 cm-1,分别对应了L 配体中的P=O 键的伸缩振动和弯曲振动、P—O—C 的伸缩振动;1 081、979、817、897 cm-1分别对应了磷钨酸中的P—O 键伸缩振动、W—Oc—W 的伸缩振动、W—Oe—W 的伸缩振动和W=O 的伸缩振动。配合物2 主要的特征吸收峰:1 208、596、1 034 cm-1,分别对应了L 配体中的P=O键的伸缩振动和弯曲振动、P—O—C 的伸缩振动;1 081、979 、820、896 cm-1分别对应了磷钨酸中的P—O键伸缩振动、W—Oc—W的伸缩振动、W—Oe—W的伸缩振动和W=O的伸缩振动。
与配体相比,配合物1、2 中的ν(P=O)分别向低频区移动32、52 cm-1,L与稀土离子的配位反应导致了特征峰红移,而1 和2 偏移程度不同,原因可能来自于配位数不同导致的不同配位环境的影响。
配合物1~2 的PXRD 表明,样品在5°~50°范围内有明显的晶相衍射峰。合成样品的主要衍射峰与模拟结果吻合较好,表明粉末样品配合物1~2 的晶相纯度较高(图S2)。
对配合物1~2 进行了TGA,如图S3 所示。配合物1 在80~150 ℃损失的重量在1.88%,此处归因于晶胞中乙腈分子的损失(计算值1.90%)。在240~330 ℃热重曲线剧烈下降7%,归因于部分配体L 的结构崩塌。330 ℃后热重曲线缓慢下降直至几乎不发生变化。配合物2 在240~330 ℃温度范围内出现剧烈的失重,损失了11.04%,归因于部分配体的结构崩塌。在330 ℃以后热重曲线缓慢下降直至几乎不发生变化。配合物1~2中的紧密堆积的三维超分子结构增强了晶体的稳定性,在260 ℃内可保持稳定。
2.3 发光性能
在室温下,对配合物1、2 和配体L 进行了固态荧光光谱测试分析(图7),将相应的激发波长和发射波长列于表3。

图7 配合物1(a)和2(b)的荧光激发和发射光谱Fig.7 Fluorescence excitation and emission spectra of complexes 1(a)and 2(b),respectively

表3 配体L和配合物1、2的荧光激发发射峰Table 3 Fluorescence excitation emission peaks of ligand L and complexes 1 and 2
在激发波长λex=307 nm 下,配体L 的发射波长为λem=361 nm,该发射峰主要归因于配体分子的n→π电子跃迁。对配合物1,在激发波长λex=393 nm 下,产生5 个f-f跃迁发射峰,显示出Eu3+特有的红光发射,波长λem分别为587、595、613、652 和699 nm,分别对应于5D0→7F0、5D0→7F1(磁偶极跃迁)、5D0→7F2(电偶极跃迁)、5D0→7F3和5D0→7F4[35],I(5D0→7F2)/I(5D0→7F1)值为1.00,5D0→7F2跃迁与5D0→7F1跃迁强度接近表明Eu3+离子处于一个高对称的微环境中,即Eu3+处在一个晶格的对称中心位置。对配合物2,在激发波长λex=466 nm 下,产生4 个f-f跃迁发射峰,显示出Tb3+特有的绿光发射,发射波长λem分别为510、542、576 和608 nm,分别对应于5D4→7FJ(J=6,5,4,3)跃迁[36],其中对应于5D4→7F6跃迁的发射峰最强。
此外,在室温下测量了配合物1、2 在固态下的发光量子产率,分别为19.43%、9.74%。因为多酸没有与稀土发生配位,所以认为光致发光的主要原因是通过配体使Ln(Ⅲ)离子敏化,L 充分吸收的光能通过分子能量传递模式把能量再传递给与之配位的中心稀土离子,即所谓的天线效应。根据文献报道,L 与稀土配位有桥联和螯合2 种方式[37],而螯合更有利于减弱配体上多条碳链的摆动和旋转,同时满足稀土离子的高配位数,配合物1 与2 都是多个配体分子形成的螯合配位方式,这可能与多酸离子起到的模板作用有关。配合物1 与2 的荧光强度存在明显差异,这是由于配合物1 有4 个配体分子螯合配位,而2 只有3 个配体配位,所以配合物1 的天线效应强于2,同时2 中1 个配位水分子导致荧光猝灭效应[38],所以2的荧光强度较弱。
2.4 太赫兹光谱
配合物1 及其原料的太赫兹光谱如图8 所示。可以清楚地观察到配体L和磷钨酸的THz-TDS吸收峰完全不同。配体L 的吸收峰强度极大,峰形在0.2~2.4 THz 内先迅速上升,然后缓慢下降,磷钨酸和配合物1 的吸收峰强度极小,峰形在0.2~2.4 THz内平缓,只显示出极多小的尖峰。对比原料与配合物1 的THz 光谱,可以发现新物质的产生伴随着新的吸收峰的出现,这归因于新分子骨架和分子间相互作用的生成。磷钨酸和配体L的一些特征峰在配合物1 的THz 光谱中消失或发生了移动,在0.38、0.44、0.73、0.91、1.01、1.75、1.90 和1.99 THz 附近的尖峰能够归属为磷钨酸的特征吸收峰;配体L 的THz 光谱可整体看作一个在1.05 THz 附近的巨大吸收峰,此吸收峰随着配位反应的发生而在配合物1中消失。
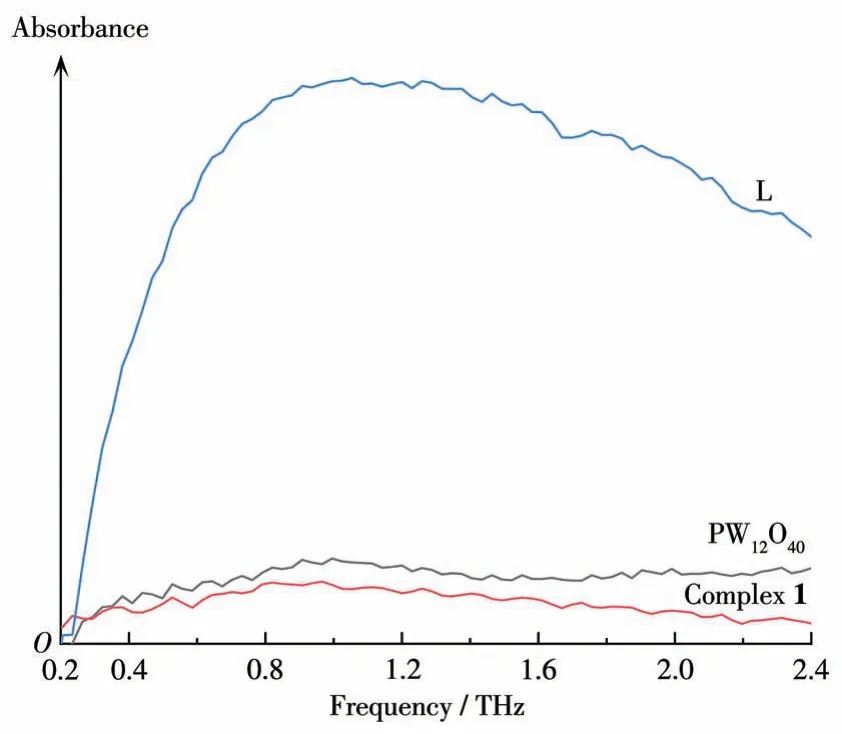
图8 配合物1及原料在0.2~2.4 THz的太赫兹光谱Fig.8 Terahertz spectra of complex 1 and raw material at 0.2-2.4 THz terahertz light
配合物1~2 的太赫兹光谱如图9 所示。配合物特征峰如下:0.23、0.35、0.53、0.70、0.82、0.96、1.11、1.26、1.41、1.55、1.72、1.85、1.99、2.13、2.31 THz(配合物1);0.26、0.43、0.52、0.61、0.70、0.81、0.87、0.99、1.25、1.34、1.43、1.55、1.63、1.72、1.87、1.96、2.13、2.28 THz(配合物2)。配合物1和2的峰形相似,有众多相同的吸收峰,这归因于相似的晶体堆积结构,太赫兹辐射具有检测分子间振动的能力,这些振动模式主要来自分子骨架振动和分子间相互作用,该特性可用于配合物的无损检测。遗憾的是,虽然太赫兹光谱可以表征弱效应,但该配合物包含复杂的分子间作用力,想要对每一个吸收峰给出正确的归属需要未来进一步的探索。
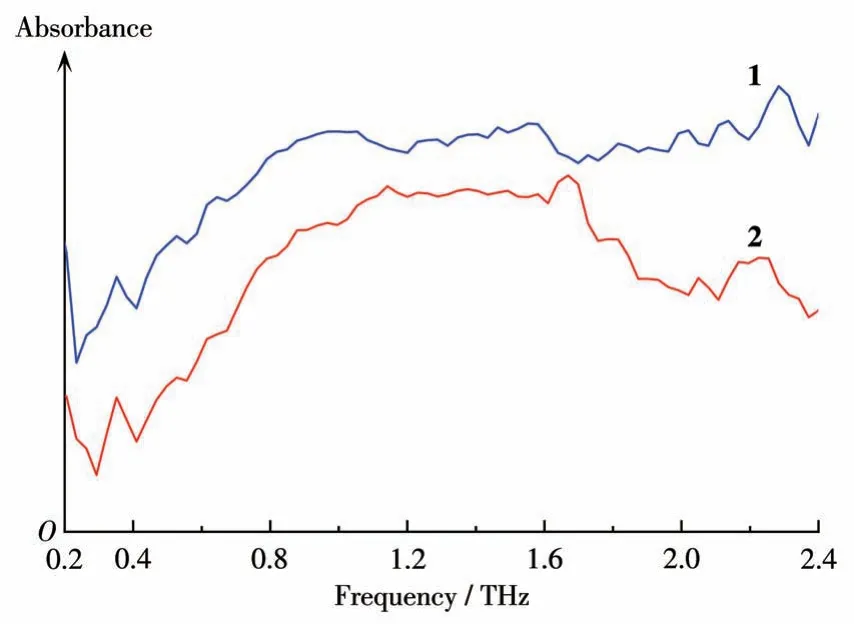
图9 配合物1~2在0.2~2.4 THz的太赫兹光谱Fig.9 Terahertz spectra of complexes 1-2 at 0.2-2.4 THz
3 结 论
合成了2种多酸基镧系双膦酸酯配合物[Eu(L)4]PW12O40·2CH3CN (1)和[Tb(L)3(H2O)]PW12O40(2),使用单晶X 射线衍射,元素分析,红外光谱,粉末X 射线衍射、热重分析和太赫兹时域光谱等对配合物的性质进行了表征,分析了配合物的晶体结构及分子间弱作用力,研究了配合物的发光等性能。配合物1和2 通过氢键构筑成三维超分子结构,配合物1 与2分别表现出典型的铕配合物红光发射和铽配合物绿光发射。2 种配合物的太赫兹时域光谱为快速检测配合物中包含的弱作用力提供了光谱学依据。
Supporting information is available at http://www.wjhxxb.cn
猜你喜欢
食品工业科技(2023年4期)2023-02-14
中国食品学报(2019年10期)2019-11-12
雷达学报(2018年1期)2018-04-04
雷达学报(2018年1期)2018-04-04
雷达学报(2018年1期)2018-04-04
中学化学(2015年12期)2016-01-19
中国洗涤用品工业(2015年2期)2015-02-28
原子与分子物理学报(2014年3期)2014-02-28
无机化学学报(2014年1期)2014-02-28
食品科学(2013年8期)2013-03-11
