
含苯并咪唑三足配体的金属配位化合物的合成及对碘的吸附性能
2024-01-20 03:55张星星牛云垠
无机化学学报 2024年1期
张星星 牛云垠
(郑州大学化学学院绿色催化中心,郑州 450001)
0 引 言
随着社会经济的迅猛发展及人口的迅速增长,人们对能源的需求量也在快速增加。然而煤、石油、天然气在使用过程中会污染环境,并且不可再生[1]。因此开发和利用低碳新能源至关重要。核能作为一种绿色、清洁、可持续的新兴能源,具有超高的能量密度以及低碳排放等优势而受到广泛关注[2]。但是发展核能也会带来潜在的风险,比如历史上发生的核泄漏事件(如1986 年切尔诺贝利核电站爆发的核事故以及2011 年福岛核泄漏事故等)[3]。在核裂变过程中会产生放射性碘(主要是129I和131I),其半衰期分别约为1570 万年和8 d[4-9]。放射性碘都具有很强的挥发性,它们可以迅速分散在空气和水中,破坏地球的生态系统,直接对人类健康构成了威胁[10-11]。放射性碘通过食物链的富集,不断进入人体[5],参与人体代谢[12],引起甲状腺损伤[12-15],致癌[16],甚至会提高胎儿畸形比例[17-18]。另外,放射性碘与挥发性有机物结合可对心肺和中枢神经系统造成不可逆的损害[19-20]。因此,如何快速高效捕获与储存核泄漏和核裂变产生的放射性碘,已经成为科学家研究的热点问题。
目前捕获碘的方法主要有膜分离法[21]、液体吸收法、空气吹出法、离子交换法[22-23]、化学沉淀法[24]、吸附法等。捕获碘技术虽然各有优势,但前面几种技术普遍生产成本高、操作复杂。相反,吸附法捕获碘具有高效、易操作、低能耗、环境可持续性等优点[25],在碘捕获中得到了广泛应用。吸附法是利用吸附剂的独特结构与吸附质的相互作用(物理或化学作用)进行的,而多孔吸附剂已经进入到最前沿的清除放射性污染物的应用中,具有高效去除的优势。传统的多孔碘吸附剂有活性炭,较高的孔隙率使其对碘有高效的物理吸附作用,但是活性炭制备过程繁杂,成本也比较高[26]。纳米金属盐与金属氧化物通常利用碘与金属离子发生反应进行碘的分离富集,但金属氧化物与水分离比较困难,操作复杂[27]。金属有机骨架(MOFs)是由配体与金属离子或金属簇连接形成的兼有柔性和刚性的多孔材料,因其可通过中心金属离子和配体种类的调控,制备出结构丰富、孔隙发达、比表面积大的骨架材料而应用到吸附分离、荧光、催化等许多领域。因此,MOFs材料吸引了各个领域科学家们的广泛兴趣并得到深入研究。Tina M.Nenoff 教授等的研究发现,2-甲基咪唑酸酯与Zn形成的沸石咪唑酸骨架ZIF-8的孔径大小与碘分子大小接近,同时配体中N 可对碘产生电子转移形成电荷转移配合物,导致其拥有较高的吸附量[28]。不同特性的配体在决定最终的骨架和对不同污染物的去除性能方面发挥着关键作用。碘是一个电子受体,与很弱的电子供给体都能形成电子转移化合物,而N原子富含电子,与碘可以发生电荷转移作用,它们之间的强相互作用使得富电的N原子更容易吸附I2[29]。综上所述,设计合成富含氮的配体及其MOFs 对高效捕获碘具有重要的理论和实践意义。
本研究选用2,2′,2″-三(1-苯并咪唑基)乙基胺(L1)配体构筑了2 种新型过渡金属配位化合物[Co3(L1)2Cl6]n(1)和{[Cu(L1)(SO4)]·2CH3OH}n(2)。化合物1 和2 具有复杂的结构,并且含氮量高,因此它们对碘都可能具有优异的捕获性能。
1 实验部分
1.1 试剂与仪器
硫酸铜、氢氧化钾、环己烷购自天津市科密欧化学试剂有限公司。苯并咪唑购自北京百灵威科技有限公司。氯化钴、二氯亚砜、三乙醇胺、碘购自麦克林。DMF、无水乙醇购自天津市富宇精细化工有限公司。三氯甲烷购自国药集团化学试剂有限公司。以上试剂均为分析纯。
傅里叶变换红外光谱(FTIR)在Bruker Vector 22 FT-IR 光谱仪上使用KBr 压片法测量,波长范围400~4 000 cm-1。单晶X 射线衍射在Bruker SMART CCD 仪器上进行测试。化合物的粉末X 射线衍射(PXRD)图是在X′Pert PRO 仪器上在室温下获得:工作电压40 kV,电流40 mA,辐射源为CuKα(λ=0.154 18 nm),以4 (°)·min-1扫描5°~50°的(2θ)范围。热重分析(TGA)数据用NETZSCHTG209 热重分析仪获得:在氮气氛围中(气体流速30 cm3·min-1),从30 ℃以5 ℃·min-1的速率升高到800 ℃,记录样品的质量损失变化。元素分析(EA)是在Perkin-Elmer 240的仪器上测定化合物中的C、H、N元素的含量。
1.2 有机中性配体L1的合成
1.2.1 中间体T1(2,2′,2″-三氯乙基胺盐酸盐)的合成
首先向250 mL 的三颈烧瓶中加入二氯亚砜(13 mL,0.175 mol)和CHCl3(20 mL),然后向其中逐滴滴加三乙醇胺(14.9 g,0.10 mol)和CHCl3(25 mL)的混合溶液,室温下滴加1 h,滴加完毕后在室温下继续反应1 h,直到气体析出完毕。然后继续升高温度达65 ℃,将混合液在该温度下加热回流4 h,冷却至室温,得到白色固体。过滤混合物,用CHCl3(3×10 mL)清洗3 遍,真空干燥,产率在30%以上[30]。反应方程式为:
具体的合成路线如图1所示:

图1 中间体T1的合成路线Fig.1 Synthesis route of intermediate T1
1.2.2 配体L1的合成
称取0.809 2 g(6.84 mmol)苯并咪唑与4 g(35.8 mmol) KOH 加入到100 mL 三颈烧瓶中,滴加5 mL DMF 到上述烧瓶中并超声溶解,将圆底烧瓶置于恒温油浴锅中。用2 mL DMF 超声溶解0.413 2 g(2.02 mmol)中间体T1 并逐滴滴加到圆底烧瓶中,混合液在60 ℃搅拌回流24 h。冷却至室温后,倒入50 mL的冰水中,出现白色固体,搅拌30 min 使KOH 充分溶解,放置2 h,白色固体沉降下来后抽滤,用冷水洗涤数次得到中性配体L1,真空干燥,产率为50%[31-32]。具体的合成路线如图2所示:
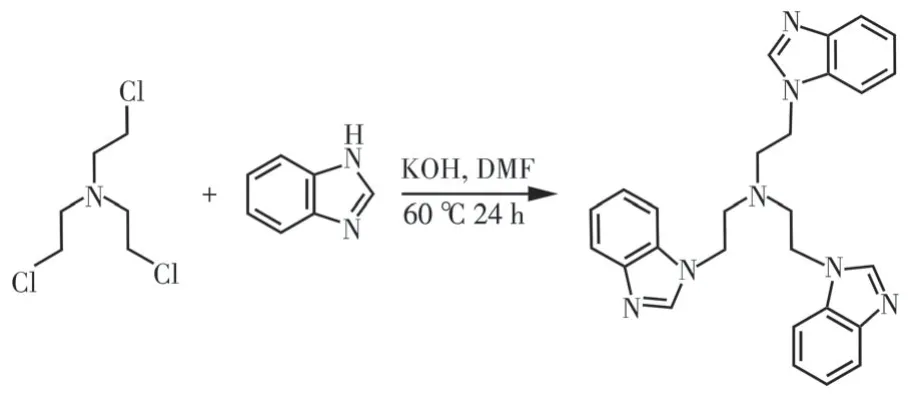
图2 配体L1的合成路线Fig.2 Synthesis route of ligand L1
1.3 化合物的合成
1.3.1 化合物1的合成
将配体L1(0.013 5 g,0.03 mmol)和CoCl2·6H2O(0.007 1 g,0.03 mmol)分别加入甲醇(4 mL),超声至完全溶解,将混合液搅拌60 min,放入25 mL 不锈钢反应釜中,置于程序控温烘箱中,30 min 内升温至120 ℃,120 ℃下恒温3 d,24 h 内降至室温,关闭烘箱,待反应釜完全冷却后在釜底和釜壁上得到蓝色块状透明晶体,产率约为54%。IR(cm-1,KBr):3 440(m),2 924(w),1 632(w),1 517(w),1 464(w),1 392(w),1 296(w),1 268(w),1 203(w),1 122(w),751(w),561(w),464(w).元素分析按C54H54Cl6Co3N14的计算值(%):C,50.29;H,4.19,N,15.21。实测值(%):C,50.16;H,4.05;N,15.33。
1.3.2 化合物2的合成
2 的合成过程与1 相似,只是用CuSO4(0.005 1 g,0.03 mmol)代替CoCl2·6H2O。待反应釜程序降温完全冷却后,在釜底和釜壁上得到蓝色长条透明晶体,产率约为62%。IR(cm-1,KBr):3 750(m),3 432(w),3 075(w),2 924(w),1 615(w),1 510(w),1 482(w),1 465(w),1 386(w),1 298(w),1 258(w),1 236(w),1 143(w),1 120(w),1 017(w),896(w),745(w),639(w),614(w),595(w),518(w),462(w)。元素分析按C29H35CuN7O6S的计算值(%):C,51.71;H,5.20;N,14.56。实测值(%):C,51.37;H,5.32;N,14.26。
1.4 化合物的晶体结构测定
通过水热法培养出单晶后,在光学显微镜下挑选出大小适宜,质地均匀,形状规则透亮的块状/粗长条晶体作为待测样品在单晶X射线衍射仪上进行测试。待测晶体利用MoKα射线(λ=0.071 073 nm)进行X射线检测和数据收集,然后进行吸收校正,并利用OLEX2 程序包进行解析[33]。所有非氢原子均采用各向异性精修方法,所有氢原子均加入到几何最优位置进行各向同性精修。化合物的晶体学参数和主要键长、键角分别列于表1和2。
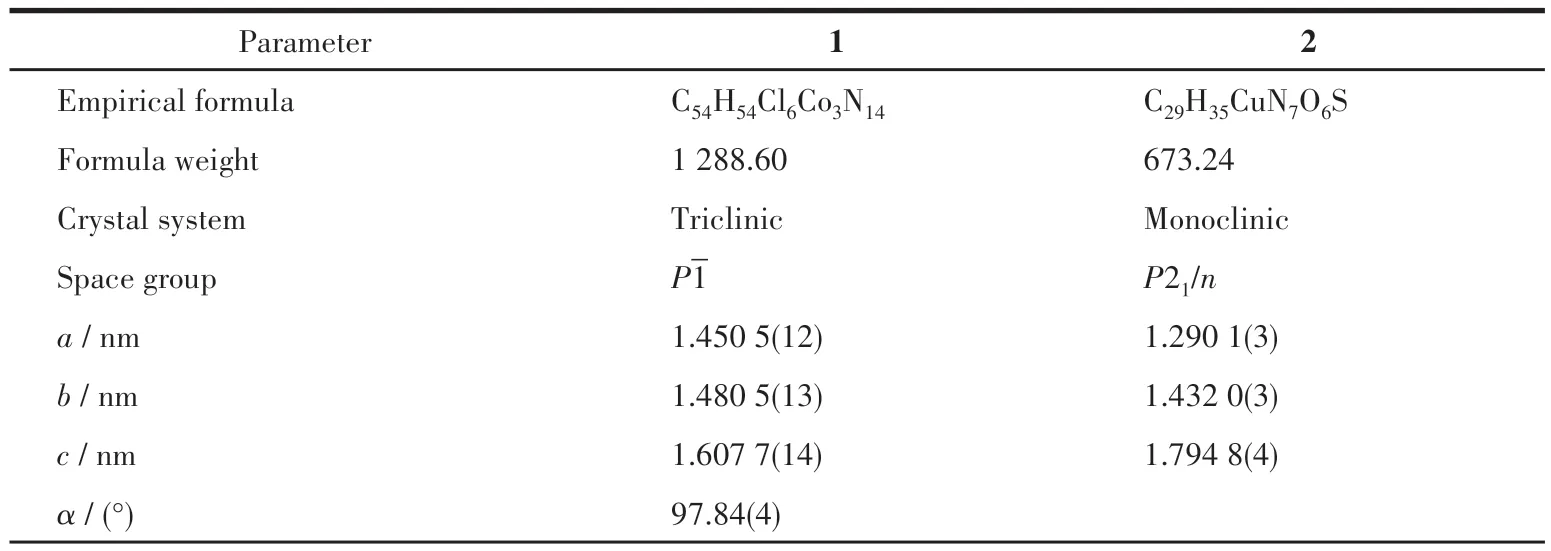
表1 化合物1和2的晶体学数据Table 1 Crystallographic data of compounds 1 and 2

表2 化合物1和2的主要键长(nm)和键角(°)Table 2 Selected bond lengths(nm)and angles(°)of compounds 1 and 2
CCDC:2288280,1;2288281,2。
1.5 吸附实验
1.5.1 化合物对环己烷溶液中碘的吸附实验
化合物1 和2 捕获环己烷溶液中碘的实验是在室温条件下进行的。具体操作过程如下:首先,配制质量浓度为500 mg·L-1的环己烷-碘溶液。称取一定量的化合物分别放到25 mL 的小玻璃瓶中,分别向小瓶中加入5 mL 上述配制的环己烷-碘溶液。在不同的时间点,用注射器抽取适量小瓶中环己烷-碘溶液,离心,取适量上清液,用紫外分光光度计在522 nm 处测量碘的吸光度。根据环己烷-碘溶液的工作曲线来确定环己烷溶液中碘的残留量。由此计算吸附剂的吸附性能,用公式1 来计算吸附剂对碘的去除率η,用公式2 来计算在t时刻吸附剂对碘的吸附容量(qt,mg·g-1)[34]:
式中,ρ0为碘的初始质量浓度(mg·L-1);ρt为t时刻碘的质量浓度(mg·L-1);V是环己烷-碘溶液的体积(mL);m是吸附剂的质量(g)。所得的结果为3次平行实验的平均值。
1.5.2 化合物对气态碘的吸附实验
在常压75 ℃条件下,研究了化合物对气态碘吸附行为。通过在不同时间点称量化合物的质量来评估对气态碘的捕获能力。具体步骤如下:准确称取50 mg 化合物,并将其置于已称重的敞口玻璃称量皿中。将盛有化合物的敞口玻璃称量皿置于带有过量碘蒸汽的密封干燥器中。将密封的干燥器转移至75 ℃的烘箱中,静置,每隔一定的时间,取出干燥器中的称量皿,待其冷却后进行称重。整个气态碘吸附实验进行了72 h,通过在不同时间称量瓶的增重确定化合物对气态碘的吸附量,并使用公式3对吸附剂吸附气态碘的能力进行了计算:
式中,q为t时刻吸附剂捕获气态碘的容量(g·g-1);m0为吸附剂在初始时刻的质量(g);mt为吸附剂在t时刻的质量(g)。所得的结果为3 次平行实验的平均值。
1.6 吸附动力学实验
为进一步探究化合物对碘的吸附机理和吸附性能,使用准一级动力学模型(公式4)和准二级动力学模型(公式5)对其进行吸附动力学的研究:
式中qe为吸附剂的平衡吸附容量(mg·g-1);k1为准一级反应速率常数(min-1);k2为准二级反应速率常数(g·mg-1·min-1)。
1.7 化合物对碘吸附的循环实验
考虑到吸附剂的经济性,需要考察吸附剂对碘吸附的循环再生性。用无水乙醇洗涤吸附碘的化合物,使其释放出吸附的碘,将释放碘之后的化合物在75 ℃的真空干燥箱中干燥,进行下一次碘吸附实验。然后重复上述洗涤-干燥-碘吸附过程4次。
2 结果与讨论
2.1 化合物的晶体结构描述
2.1.1 化合物1的晶体结构描述
单晶X 射线衍射分析表明,1 属于三斜晶系P1空间群。如图3a 所示,化合物1 的最小结构单元是由2 个中性配体L1 和3 个[CoCl2]连接而成的三核结构。3个Co(Ⅱ)离子的配位环境相同,都是四配位,由2个L1配体中的N 原子和2个Cl原子配位形成四面体构型。主要键长范围:Co—Cl 0.222 4(4)~0.225 1(3)nm,Co—N 0.200 2(6)~0.204 2(6)nm;主要键角范围:∠Cl—Co—Cl=107.42(16)° ~116.90(6)° ,∠Cl—Co—N=101.55(19)°~114.30(19)°,∠N—Co—N=103.9(3)°~109.4(2)°。如图3b 所示,每相邻2 个配体L1 中的3个N 原子分别与3 个Co 原子相连形成一维链状结构,其中相邻配体L1 中2 个N 原子通过与2 个Co 原子相连围成一个闭合的方形结构,其相距最远的距离是1.263 4 nm,最近的距离是0.415 2 nm,图3c 为1的堆积图,显示其形成了三维网状超分子结构。

图3 (a)化合物1的不对称结构单元;(b)1的链状结构;(c)1的堆积图Fig.3 (a)Asymmetric structural unit of compound 1;(b)Chain structure of 1;(c)Stacking diagram of 1
2.1.2 化合物2的晶体结构描述
单晶X 射线衍射分析表明,2 为单斜晶系,空间群为P21/n。如图4a 所示,2 的最小不对称结构单元是由1个中性配体L1、1个[CuSO4]单元和2个游离的CH3OH分子组成。Cu2+离子为三配位,与1个N原子和2 个氧原子形成四面体几何构型。主要键长:Cu—O 0.199 7(2) nm,Cu—N 0.200 5(3) nm;主要键角范围:∠O—Cu1—N=92.64(9)°~163.19(10)°,∠N—Cu1—N=87.89(10)°~166.21(10)°。化合物2 的晶胞骨架结构(图4b)由6 个配体和6 个[Cu2SO5N]结构单元相连而成,结构中心为一个孔洞结构。骨架呈中心对称,其中1 个Cu2+离子与3 个N 和3 个O 连接在一起,呈现八面体的结构单元,Cu 与O 之间的距离为0.256 5、0.199 7 和0.256 5 nm;另外1 个Cu2+离子和2 个氧连接在一起,Cu 与O 之间的距离为0.199 7和0.256 5 nm,2 个Cu2+与2 个O 原子组成了一个平行四边形的结构。化合物2的晶胞骨架结构沿a、b、c方向不断伸展得到了三维立体结构,图4c 为a轴方向的网状结构,形成了圆环形的孔洞结构相互交错。图4d为b轴方向的网状结构,2个Cu2+离子与其他原子组成的结构单元排列成相互平行的结构,中间由2种不同的孔洞结构相互交错而成。
2.2 化合物的表征
2.2.1 化合物的红外光谱和PXRD
图5a 和5b 为化合物1 和2 的红外光谱图,以1 的红外光谱图为例,3 441 cm-1对应的是苯并咪唑上—N—H 的伸缩振动。1 632 cm-1为苯并咪唑上—C=N—的伸缩振动峰。1 632、1 517 和1 464 cm-1吸收峰为苯环骨架振动产生的,说明该结构中含有苯环。1 269 cm-1为C—N键的伸缩振动。

图5 化合物1(a)和2(b)的红外光谱图;1(c)和2(d)的PXRD图Fig.5 (a)IR spectra of compounds 1(a)and 2(b);PXRD patterns of 1(c)and 2(d)
化合物1 和2 的PXRD 数据如图5c 和5d 所示,实验数据和模拟数据吻合度较高,说明化合物1和2具有较高的相纯度。
2.2.2 化合物的稳定性
我们通过TGA 测得了化合物1 和2 的TG 曲线以研究其热稳定性。如图6a 和6b 所示。化合物在加热后会有多步分解过程。化合物1 在30~350 ℃未见明显失重,说明化合物1具有良好的耐热性,当温度达到350 ℃时,化合物1 开始失重,期间骨架开始坍塌,当温度加热到460 ℃时化合物1 的失重为50.27%,这归因于配体L1 的分解。460 ℃后继续升温,化合物1 出现缓慢失重,一直到800 ℃,失重为16.37%,460~800 ℃期间的失重归因于无机金属盐的分解。化合物2 在285 ℃之前具有良好的耐热性,期间有轻微的失重(9.48%),归因于游离的CH3OH 溶剂分子的失去。285 ℃后继续加热,化合物2 出现大幅度失重,说明其骨架坍塌,在420 ℃化合物2 失重达到稳定状态,285~420 ℃的失重达到60.18%,归因于化合物2 中配体分子与CuSO4分子的分解。化合物在有机溶剂中也表现出特定的稳定性。将化合物1 和2 在不同的有机溶剂中浸泡1周,之后对浸泡前后的材料进行红外和PXRD 的测试分析,化合物1和2的红外谱图如图6c和6d所示,PXRD 图如图6e 和6f 所示。由于化合物1 和2 的红外谱图和PXRD 图的峰形没有明显变化,可确定它们在浸泡后仍保持原来的晶体结构,这表明2 种化合物具有良好的溶剂稳定性。
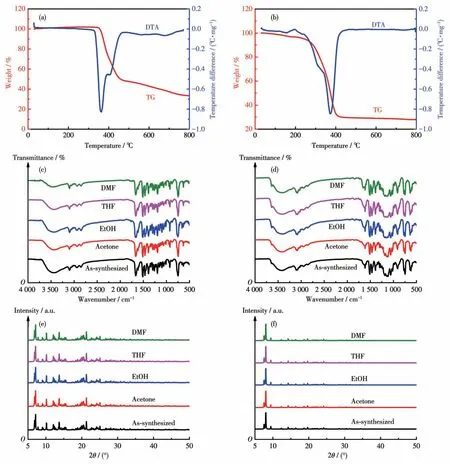
图6 化合物1(a)和2(b)的TG-DTA曲线;化合物1(c)和2(d)在不同有机溶剂中浸泡7 d前后的红外光谱对比图;化合物1(e)和2(f)在不同有机溶剂中浸泡7 d前后的PXRD对比图Fig.6 TG-DTA curves of compounds 1(a)and 2(b);Comparison of IR spectra of 1(c)and 2(d)before and after being soaked in different organic solvents for 7 d;Comparison of PXRD patterns of 1(e)and 2(f)before and after being soaked in different organic solvents for 7 d
2.2.3 化合物的N2吸附-脱附
为了研究化合物的吸附性能,进行了N2吸附-脱附实验,对化合物的孔径分布进行了表征。测试结果如图7a 和7b 所示,化合物1 和2 均呈现出典型的Ⅱ型吸附等温线,化合物1 和2 的孔体积分别是6和19 cm3·g-1。通过Olex2 对化合物的结构进行计算,得出化合物1 和2 的孔洞率分别为7.1%和26.3%。初步分析表明化合物2 比化合物1 有更优的吸附性能,而实际的吸附效果需要吸附实验进一步的验证。

图7 化合物1(a)和2(b)的N2吸附-脱附等温线Fig.7 N2 adsorption-desorption isotherms of compounds 1(a)and 2(b)
2.3 化合物对环己烷中碘的吸附性能
2.3.1 吸附剂用量对碘吸附的影响
为了研究吸附剂对环己烷溶液中碘的最佳吸附效果,探索了不同吸附剂的量对碘的吸附实验。以化合物1 为例,分别准确称取了10、20、40、70 mg的化合物1 于小玻璃瓶中,各加入5 mL 环己烷-碘溶液,每隔一定时间段测量碘溶液的吸光度,测试结果如图8a~8d 所示。通过实验结果的对比发现70 mg 化合物1 对碘的吸附效果最好。不同吸附剂量对应的去除率分别为82.05%、88.88%、95.42%和96.68%(图8e)。达到吸附平衡时不同吸附剂量对应的吸附容量分别为188.66、102.87、55.52 和32.18 mg·g-1(图8f)。化合物1 对碘的吸附可以分成3 个阶段,第一个阶段为7 h 之前,在此吸附过程中,化合物表面有大量的活性位点,大量的碘快速扩散到化合物的表面,导致碘被快速吸附。第二阶段,随着吸附时间增长,化合物中的活性位点被大量的碘占据,导致化合物对碘的吸附速率变缓慢。第三个阶段,在吸附时间达到24 h 时,化合物对碘的吸附过程达到平衡,此时活性吸附位点几乎都被碘占据。在捕获碘的过程中,化合物1的颜色发生了变化,由蓝色逐渐变成墨绿色(图8f 插图),这也表明碘已经完全扩散到化合物中。化合物2对碘的吸附实验结果如图8g~8j 所示,可以发现70 mg 化合物2 对碘的吸附效果最好。不同吸附剂量对应的去除率分别为92.4%、95.28%、97.41%和98.43%(图8k)。达到吸附平衡时不同吸附剂量对应的吸附容量分别为230.99、119.10、60.88 和35.15 mg·g-1(图8l)。化合物2 对碘的吸附也可以分成3 个阶段,第一个阶段为7 h 之前,碘主要吸附在化合物表面的活性位点上,随着时间的增长,在第二阶段碘逐渐扩散到化合物的孔洞里,吸附速率变得相对缓慢,直到48 h 时到达第三阶段,化合物2 对碘的吸附达到了平衡。在捕获碘的过程中,化合物2的颜色发生了变化,由湛蓝色变成碧绿色(图8l 插图),这也表明碘已经扩散到化合物中。
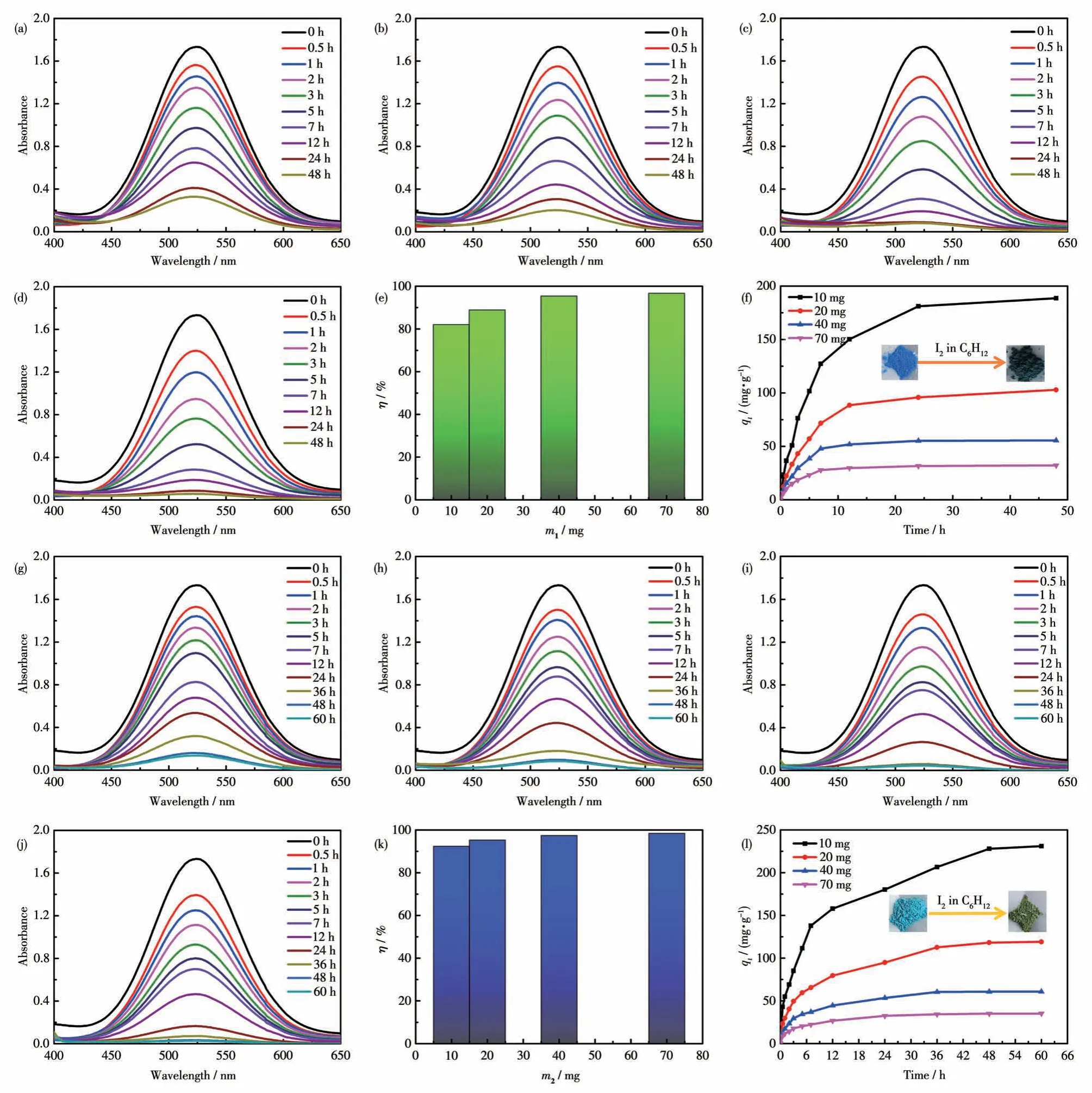
图8 (a)10 mg、(b)20 mg、(c)40 mg和(d)70 mg化合物1对碘的吸附;(e)化合物1对碘的去除率;(f)化合物1对碘的吸附容量(插图:化合物1在吸附碘前后的颜色变化);(g)10 mg、(h)20 mg、(i)40 mg和(j)70 mg化合物2对碘的吸附;(k)化合物2对碘的去除率;(l)化合物2对碘的吸附容量(插图:化合物2在吸附碘前后的颜色变化)Fig.8 Adsorption of iodine by(a)10 mg,(b)20 mg,(c)40 mg,and(d)70 mg of compound 1;(e)Removal rate of iodine by compound 1;(f)Iodine adsorption capacity of compound 1(Inset:the color change of compound 1 before and after iodine adsorption);Adsorption of iodine by(g)10 mg,(h)20 mg,(i)40 mg,and(j)70 mg of compound 2;(k)Removal rate of iodine by compound 2;(l)Adsorption capacity of compound 2 for iodine(Inset:the color change of compound 2 before and after iodine adsorption)
2.3.2 吸附动力学研究
准一级吸附动力学是假定吸附质的吸附过程受扩散步骤所控制,准二级吸附动力学是假定吸附质的吸附过程受化学吸附所控制。化合物的吸附动力学研究如图9a~9d所示,拟合的相关数据如表3和4 所示。化合物1 和2 的准二级动力学拟合的R2都在0.99 以上,并且都大于准一级动力学拟合的R2。这说明化合物1 和2 对碘的拟合吸附过程都符合准二级动力学,说明化合物对碘吸附是化学吸附过程,这归因于化合物中的富电子N 和电子受体I2发生了强的相互作用。
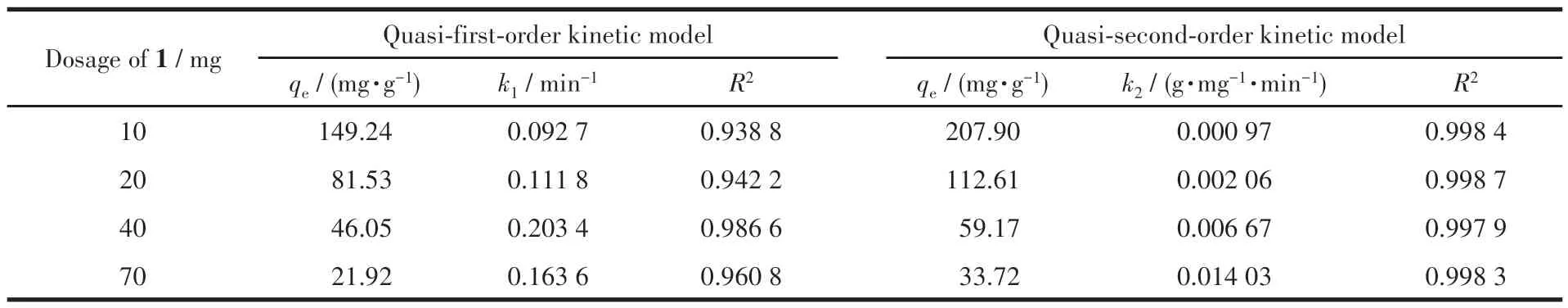
表3 化合物1对碘吸附的动力学数据Table 3 Kinetic data of iodine adsorption by compound 1

表4 化合物2对碘吸附的动力学数据Table 4 Kinetic data iodine adsorption by compound 2
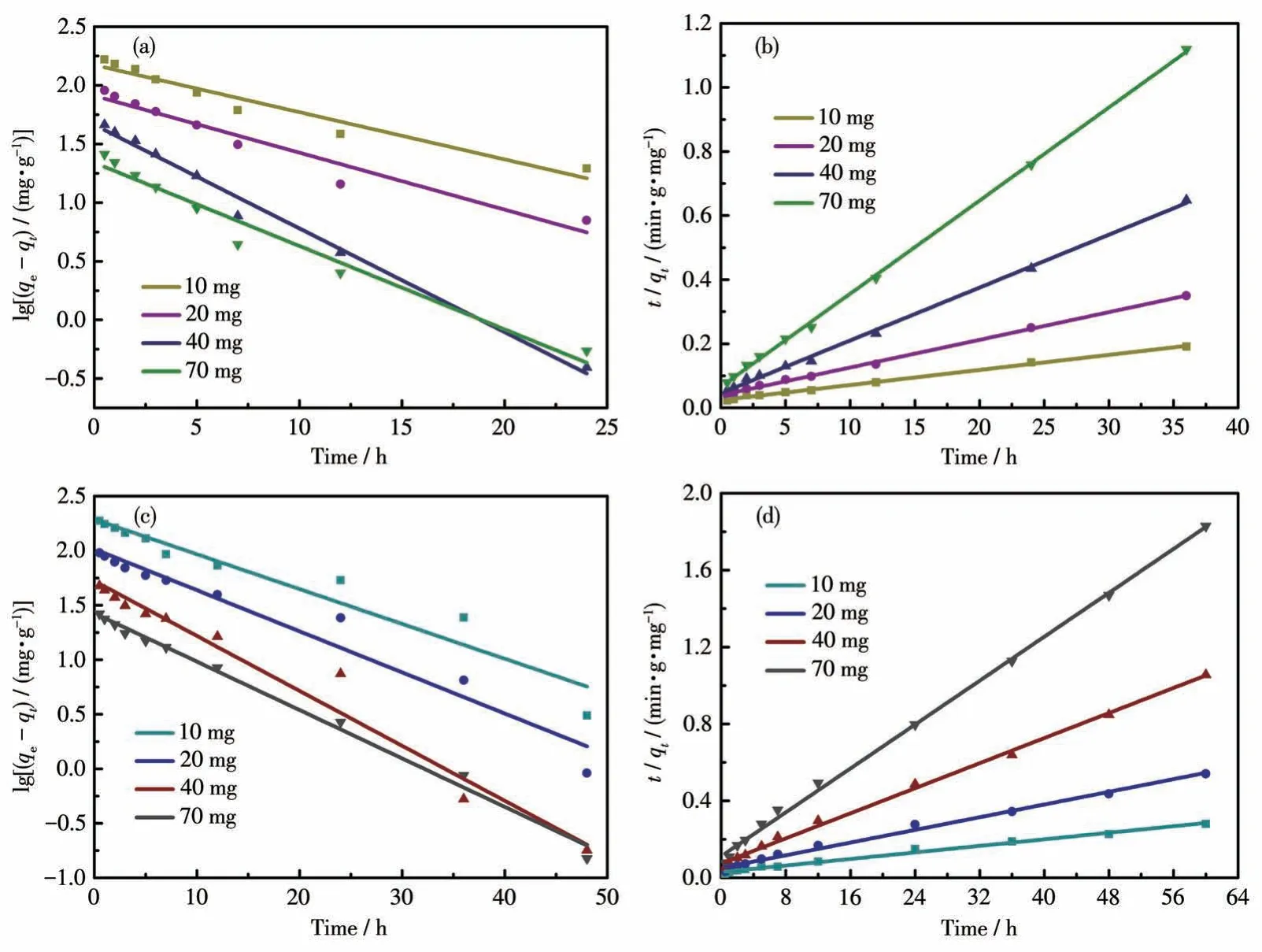
图9 化合物1对碘吸附的准一级动力学(a)和准二级动力学(b)拟合;化合物2对碘吸附的准一级动力学(c)和准二级动力学(d)拟合Fig.9 Fitting of quasi-first-order kinetics(a)and quasi-second-order kinetics(b)of iodine adsorption by compound 1;Fitting of quasi-first-order kinetics(c)and quasi-second-order kinetics(d)of iodine adsorption by compound 2
2.3.3 化合物对碘吸附的循环再生性
吸附剂的循环再生性是评估其是否具有实际应用价值的重要指标。理想的吸附剂应该具备优异的吸附性能和良好的再生性能。基于此,对化合物进行了循环再生的实验,实验结果如图10 所示,化合物1 经过了4 次循环实验之后,对碘的去除率仍然在92%以上;化合物2 经过了4 次循环实验之后,对碘的去除率仍然在97%以上,化合物1 和2 有很好的循环再生性,且化合物2 对碘的循环吸附效果比化合物1更优异。
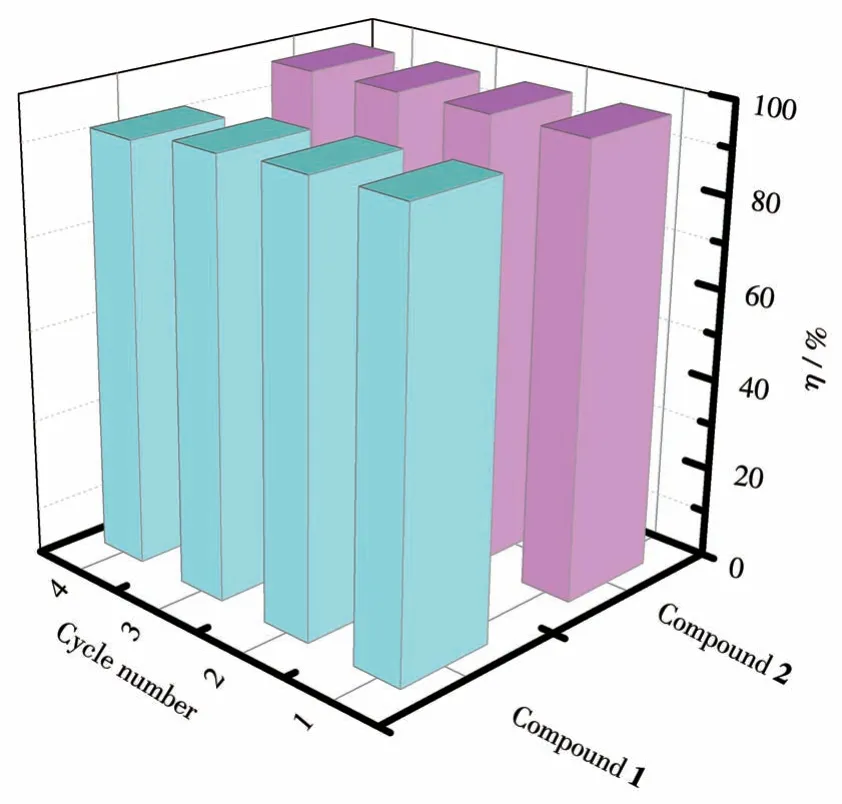
图10 化合物1和2四次循环的吸附效率图Fig.10 Adsorption efficiency diagram of compounds 1 and 2 for four cycles
2.4 化合物对气态碘的吸附性能
考察了化合物对气态碘的吸附能力。本研究中,采用重量法对不同时间间隔的化合物的质量进行测量,并根据化合物的质量变化绘制成了碘吸附曲线。如图11a 和11b 所示,化合物1 和2 均显示出较高的碘吸附性能。达到吸附平衡时,化合物1和2的碘吸附容量分别高达3.73 和3.75 g·g-1。当吸附完成后,化合物的颜色从蓝色或湛蓝色变为黑色(图11a 和11b 中插图)。吸附过程的初始阶段,化合物对气态碘的吸附量迅速增加,在36 h 之后,化合物对碘的吸附速率逐渐减慢并趋于平缓。这一实验证实化合物1和2都具有很高的气态碘捕获能力。
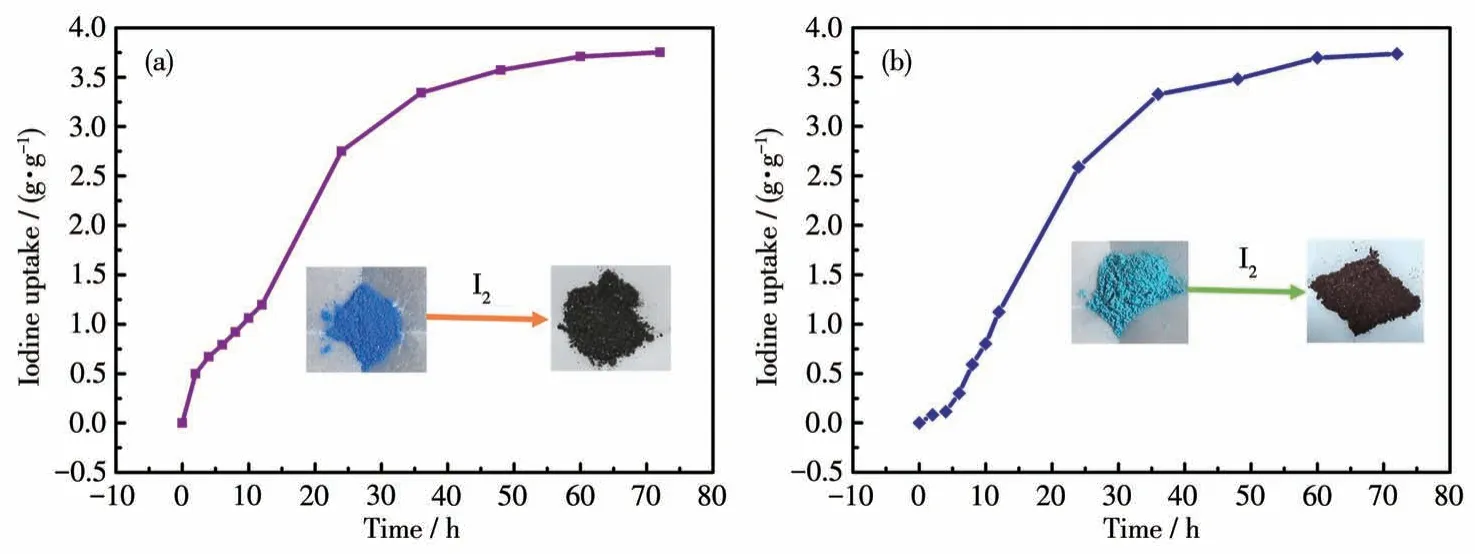
图11 化合物1(a)和2(b)对气态碘的捕获性能Fig.11 Trapping properties of compounds 1(a)and 2(b)for gaseous iodine
3 结 论
我们利用中性三足有机配体通过溶剂热法分别与氯化钴和硫酸铜反应合成了2种结构新颖的金属配位化合物1 和2,并对它们进行了IR、TG、XRD、N2吸附-脱附等表征。化合物1是一维链状结构,化合物2 是三维结构。通过实验发现化合物1 和2 对环己烷中碘有优异的吸附效果,70 mg化合物1对环己烷中碘的去除率高达96.68%,70 mg化合物2对环己烷中碘的去除率高达98.43%。此外,经过4 次循环后,化合物1对碘的去除率仍然在92%以上,化合物2 对碘的去除率仍然在97%以上,化合物的循环再生性优异。2 种化合物对碘的吸附都符合准二级动力学,为化学吸附过程。化合物1和化合物2对气态碘的吸附容量分别达到3.73和3.75 g·g-1。足以说明化合物1和2是具有良好潜能的碘捕获材料。
猜你喜欢
化工管理(2021年7期)2021-05-13
中国特种设备安全(2020年11期)2020-06-09
环境保护与循环经济(2020年4期)2020-06-08
中国特种设备安全(2019年1期)2019-03-13
柴油机设计与制造(2018年3期)2018-10-13
柴油机设计与制造(2018年2期)2018-08-29
柴油机设计与制造(2018年1期)2018-04-20
材料科学与工程学报(2016年4期)2017-01-15
合成化学(2015年4期)2016-01-17
无机化学学报(2014年6期)2014-02-28
