
噬血细胞综合征1例报道并文献复习
2024-01-18 01:41蓝方权洪峰周璇
右江医学 2023年12期
蓝方权,洪峰,周璇
(1.安徽中医药大学研究生院,安徽合肥230012;2.安徽中医药大学第一附属医院综合ICU,安徽合肥230011)
噬血细胞综合征(hemophagocytic syndrome, HPS)又叫噬血细胞性淋巴组织细胞增生症(hemophagocytic lymphohistiocytosis, HLH),是一种由淋巴细胞和巨噬细胞过度活化,损及多个脏器甚至危及生命的过度炎症反应综合征,即使规范治疗,死亡率仍极高[1]。本病病情发展迅速,可由多种病因引起,其中感染相关性HPS症状与脓毒症及全身炎症反应综合征具有相类似特征,导致诊断难度极大,易漏诊误诊,因此及早诊断并适时治疗对提高HPS患者存活率极为重要[2]。本文就我院综合ICU于2022年4月23日收治的1例老年男性HPS病例进行报道如下。
1 病例介绍
1.1 既往资料患者,男,69岁,分别在2021年8月、2021年12月无明显诱因下出现反复发热,体温最高达40 ℃,反复就诊于外院。2022年3月10日再次出现发热,于当地治疗无效后转至上级医院治疗,考虑肺部感染,治疗后症状未见好转。遂于2022年4月6日转至另一家医院治疗,完善结核斑点试验、结核Xpert、痰液结核菌培养等检查,排除活动性肺结核后予相关处理,体温控制仍欠佳。于2022年4月13日再次转至第三家医院,完善PET/CT及相关检查未发现肿瘤病变(见图1),全血宏基因组二代测序(mNGS)提示奥斯陆莫拉氏菌(序列数1),予抗感染等治疗后,效果仍不佳,10天后患者及家属要求出院。
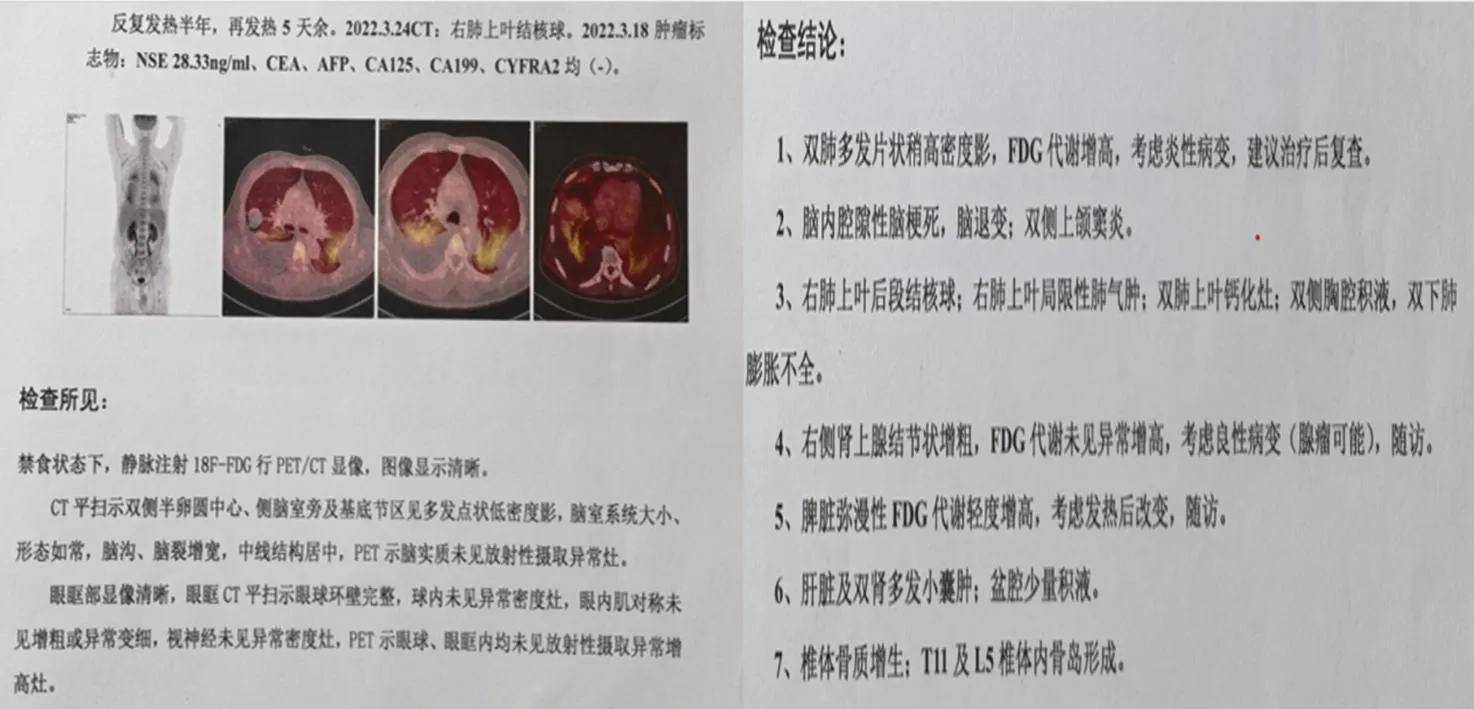
图1 PET/CT检查结果
1.2 本次入院检查资料患者出院2小时后再次呼吸困难伴意识模糊、烦躁及恶心干呕,医疗救护120接至我院急诊科,心电监护提示指脉氧60%~70%波动,血压不能维持,最低至80/40 mmHg(1 mmHg=0.133 kPa),心率达150次/分,给予气管插管后接呼吸机辅助呼吸,排除新型冠状病毒感染后,转我科进一步诊治。转入查体:T 38.8 ℃,P 120次/min,R 25次/min,BP 110/87 mmHg,意识模糊,急病面容,查体不合作;双瞳等圆,直径约3 mm,光敏;双肺呼吸音稍低,可闻及少许干湿性啰音;心率120次/分,律齐;双下肢中度凹陷性水肿。辅助检查:血气分析(机械通气后)示乳酸1.7 mmol/L,二氧化碳分压30.3 mmHg,pH7.48,氧分压117 mmHg;血常规示白细胞计数(WBC)2.77×109/L↓,血红蛋白(HGB)70 g/L↓,血小板计数101×109/L,红细胞计数2.51×1012/L↓;血生化示白蛋白25 g/L↓,降钙素原(PCT)7.117 ng/mL,C反应蛋白95.74 mg/L↑,血清淀粉样蛋白A152.76 mg/L↑;胸部CT示两肺炎症及陈旧灶,右肺局限性肺气肿;双侧肺气肿伴节段性肺不张;心包积液(见图2)。初步诊断:(1)重症肺炎;(2)脓毒性休克;(3)低蛋白血症;(4)中度贫血。
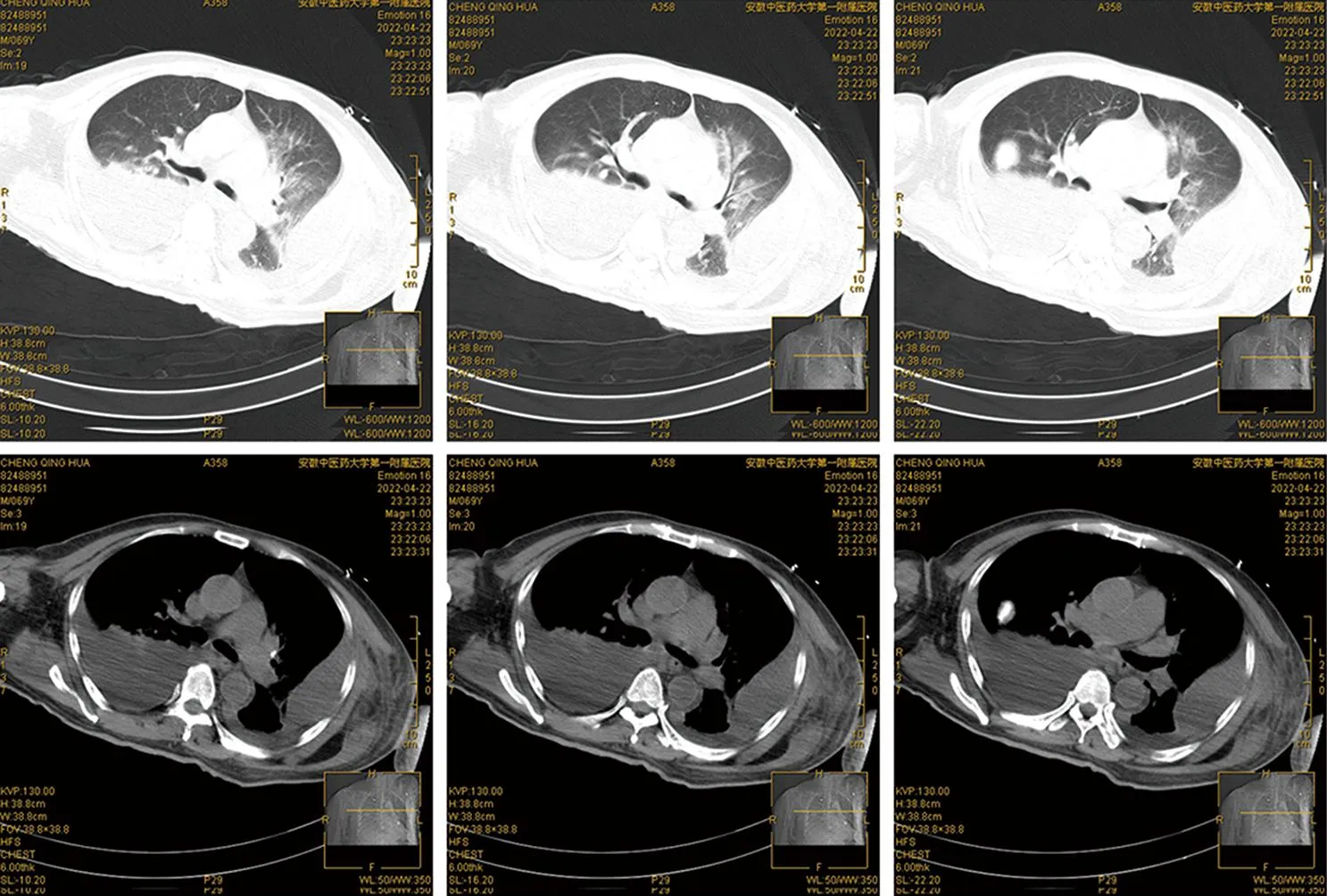
图2 入院胸部CT影像
1.3 4月23日—25日初始治疗经过入科后予抗细菌感染、机械通气、扩容补液、维持血压、营养支持等对症治疗。同时予以完善风湿系列、抗中性粒细胞胞浆抗体谱(ANCA)及自身免疫抗体等检查,均未见异常。行纤维支气管镜检查,灌洗液mNGS示(见图3):白念珠菌(序列数35)、肺炎克雷伯菌(序列数14)。结合药敏试验予以加用伏立康唑抗真菌治疗,经治疗后,患者体温高峰逐步降低,炎症指标逐渐下降,循环趋于稳定,血管活性药物去甲肾上腺素逐步减撤,但患者血红蛋白及白细胞仍持续下降,活化部分凝血酶时间(APTT)逐渐延长。
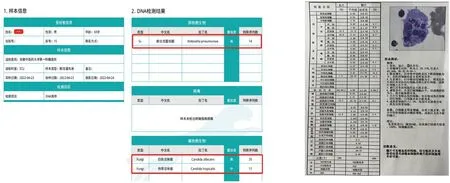
图3 肺泡灌洗液mNGS检查结果 图4 骨髓穿刺检查结果
1.4 4月26日—5月6日再次诊治过程结合患者反复发热病史、反复查阅患者相关检查,发现患者铁蛋白7146.37 ng/mL↑,甘油三酯3.3 mmol/L↑,不排除噬血细胞综合征可能。予以外送检查NK细胞活性,结果显示为1.8%↓,sCD25可溶性白介素2受体8570 U/mL↑,高度可疑噬血细胞综合征。遂予以完善骨髓穿刺,骨髓细胞检查提示(见图4):骨髓增生活跃;粒系增生,粒系占38.35%,红系占48.06%,粒∶红=0.8∶1;髓系增生,以中性中幼粒及以下阶段细胞为主,各阶段细胞形态未见异常;红系增生,以中晚幼红细胞为主;淋巴及单核细胞形态大致正常;全片可见噬血组织细胞,有吞噬成熟红细胞、幼红细胞、白细胞和血小板现象。结合临床,补充诊断:噬血细胞综合征。治疗上加用人免疫球蛋白联合甲泼尼龙冲击治疗,并予输注红悬液、血浆等对症治疗。患者一般情况逐渐好转,体温高峰降至正常范围,血三系、凝血功能逐渐好转,炎症指标进一步下降,全身水肿减轻。并于4月28日脱机拔管,5月6日复查肺部CT炎症明显改善(见图5),患者症状基本好转,予办理出院。
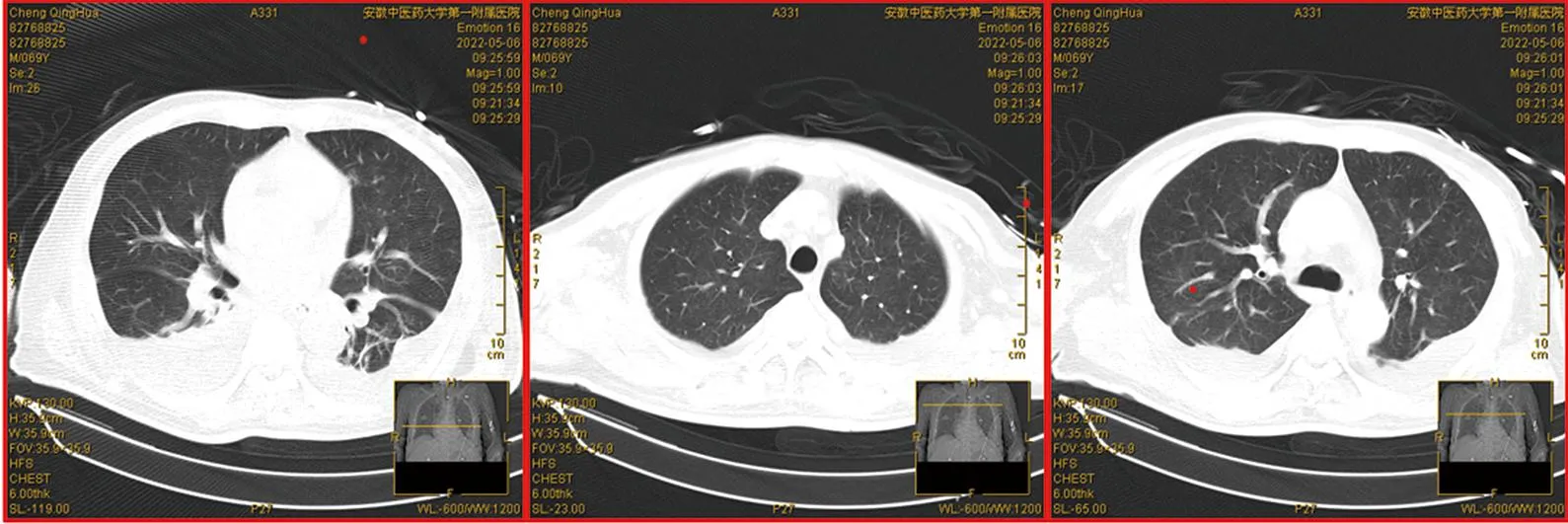
图5 出院前胸部CT影像
2 讨 论
HPS并不是一种单一的疾患,而是一种危及生命的免疫异常激活综合征,是一种罕见病,主要特点是长期发热、脾大及三系血细胞减少[3]。一项调查显示我国HPS总发病率为1.04/10万,其中儿童(<18岁)为HPS主要发病群体,约占65.40%,成年人则占34.60%[4]。HPS目前认为一般分为原发性(遗传性)及继发性(获得性)两大类,原发性HPS一般与基因缺陷及免疫缺陷综合征相关,前者可由一种常染色体或性染色体遗传至下一代,明确的致病基因为PRF1基因、Munc13-4基因及STX11等[5]。继发性HPS多由感染、自身性免疫疾病或恶性肿瘤等引起,其中感染相关性HPS与细菌、病毒、真菌及寄生虫等有关,又以EB病毒引起的最为常见,且其病死率亦是最高[6]。本例患者EB病毒核抗原IgG抗体(EBV-NAIgG)及EB病毒衣壳抗原IgG抗体(EBV-CAIgG)均阳性,考虑患者既往曾经感染过EB病毒,但已好转,结合检查考虑与本次HPS相关性不大,治疗上未针对性选择抗EB病毒类药物及其他抗病毒药物。
HPS诊断难度极大,曾一度被误认为是一种幼儿疾病,随着检测水平提升,现成人病例报道逐年增多[7]。目前国内仍以国际组织细胞协会HPS-2004方案为诊断依据(符合以下标准中的1条则诊断成立): (1)分子生物学符合HPS。(2)符合以下8条中的5条:①发热;②脾大;③全血细胞减少,累及≥2个细胞系,血红蛋白<90 g/L,婴儿(<4周)血红蛋白<100 g/L,血小板<100×109/L,中性粒细胞<1.0×109/L;④高甘油三酯血症和(或)低纤维蛋白原血症,甘油三酯≥3.0 mmol/L(或≥265 mg/L),纤维蛋白原≤1.5 g/L;⑤骨髓、脾或淋巴结活检吞噬血细胞现象,无恶性疾病证据;⑥NK细胞活性减低或缺乏;⑦铁蛋白≥500 mg/mL;⑧可溶性CD25 (或者可溶性IL-2受体)≥2400 U/mL[8]。由于本院检验水平限制,未做分子生物学方面检查,但本病例仍然符合第2条标准中的7条,故可诊断为HPS。
目前针对HPS的治疗,大体可分为诱导治疗、CNS-HPS治疗、挽救治疗、维持治疗、异基因造血干细胞移植及支持治疗等方式,针对感染相关性HPS以抗感染为主[4]。该患者以“发热待查”入住我科,情况复杂,病因不明,前期考虑重症肺炎及脓毒症相关感染性发热。积极予对症治疗,患者体温峰值虽有下降,但并未能降至正常水平,全血细胞及凝血功能仍持续恶化,随时有死亡风险。在及时调整诊断及治疗思路,完善相关检查后,考虑为HPS,与细菌、真菌感染相关可能性大,及时予糖皮质激素联合人免疫球蛋白冲击治疗,并联合抗真菌药物解除病因及加强支持治疗,患者病情逐渐好转出院,随访半年病情未再复发。
感染相关性HPS早期并无典型症状,其需与普通感染、肿瘤及自身免疫疾病等相鉴别[9]。当发现患者发热、血二系或三系下降、脾肿大、铁蛋白异常及合并感染时,应高度警惕感染相关性HPS。本病早期除对症治疗外,应以抗感染控制炎症风暴为主,还需加用免疫调节相关性药物,减缓HPS进展,阻止巨噬细胞的再度活化,同时还应关注患者基础疾病及并发症,方可改善预后。本文通过我院1例HPS报道并文献复习,希望可为各同仁提供诊断及治疗思路,提高本病诊断率及治愈率。
猜你喜欢
中国临床医学影像杂志(2022年5期)2022-07-26
天津医科大学学报(2021年4期)2021-08-21
感染、炎症、修复(2021年1期)2021-07-28
中医眼耳鼻喉杂志(2021年2期)2021-07-21
儿童故事画报(2020年12期)2020-06-23
河南科学(2020年3期)2020-06-02
云南医药(2019年3期)2019-07-25
猪业科学(2018年8期)2018-09-28
铜仁学院学报(2018年6期)2018-07-05
感染、炎症、修复(2016年4期)2016-04-17
