
猪源致病性大肠杆菌基因组比较与毒力因子分析
2024-01-12 12:41吴莉丹冉雪琴牛熙黄世会李升王嘉福
生物技术通报 2023年12期
吴莉丹 冉雪琴 牛熙 黄世会 李升 王嘉福
(1. 贵州大学生命科学学院 农业生物工程研究院 山地植物资源保护与种质创新教育部重点实验室,贵阳 550025;2. 贵州大学动物科学学院,高原山地动物遗传育种与繁殖教育部重点实验室,贵阳 550025)
大肠杆菌(Escherichia coli)是肠杆菌科埃希氏菌属的革兰氏阴性菌,是动物肠道最主要的兼性厌氧菌,通常对宿主无害。由于大肠杆菌基因组具有高度的可塑性,通过遗传变异可能获得和丢失一些基因[1],导致特定的血清型对动物致病,猪尤其易感[2]。猪大肠杆菌病在全球各地普遍存在,各年龄段均可发病,尤其是新生幼猪最易感,常给猪场带来极大的经济损失。猪源致病性大肠杆菌主要的毒力因子包括黏附系统、铁吸收和转运系统、分泌系统和毒素等[3],多种毒力因子相互协调作用,决定疾病的发生和发展进程。猪源致病性大肠杆菌首先通过黏附系统和铁吸收转运系统,黏附并定植于猪的小肠腔内表面,大量繁殖并产生毒素,再通过分泌系统将毒素分泌到细菌外,毒素被宿主吸收,损伤胃肠道血管和皮下组织,导致黄白痢、水肿病[4]。猪源致病性大肠杆菌的毒力不同,临床症状轻重不一[5]。
毒力强弱与毒力因子的数量、结构和功能密切相关。采用比较基因组学方法,比较多杀性巴氏杆菌(Pasteurella multocida, PM)的强毒株DY120818与4 株弱毒株,表明毒力的差异可能与整合结合元件ICEPmu2 有关[6];猪源肠外致病性大肠杆菌强毒菌株PCN033 和弱毒菌株PCN061 的基因序列分析提示,强毒菌株基因组中特有水平转移DNAs、特有编码黏附素、脂多糖等毒力因子基因,且拥有较多的前噬菌体序列[7]。为了系统解析本地流行的致猪腹泻大肠杆菌的致病机制,本研究以不同毒力的6 株猪源致病性大肠杆菌为研究材料,通过全基因组测序,挖掘基因组中致病相关的毒力因子基因,分析毒力因子基因发生的变异,为大肠杆菌病的诊断和防治提供理论依据。
1 材料与方法
1.1 材料
大肠杆菌弱毒菌株P32、P111 和中等毒菌株P555、P211,由本实验室从贵州某猪场腹泻仔猪粪样中分离获得[8];强毒菌株S10670(菌种编号:CICC 10670)、E24190( 菌 种 编 号:CICC 24190)购自中国工业微生物菌种保藏管理中心。
1.2 方法
1.2.1 序列测定 应用LB 培养基培养各株细菌,离心收集菌体,利用天根DNA 试剂盒提取细菌基因组DNA,构建DNA 文库,应用Illumina Hiseq 2500 测序平台(华大基因)进行基因组测序(双端,2×150bp)。此外,从NCBI 数据库下载E. coli. str.K‑12 的基因组序列作为参考。
1.2.2 基因组序列组装与基因注释 利用软件FastQC[9]评估原始测序数据的质量,以软件FastP[10]对原始测序数据中的接头和低质量reads 进行过滤;通过Spades[11]软件对过滤后的数据进行组装,GapClose[12]软件对未知碱基进行填充;采用SeqKit[13]软件过滤掉组装序列中小于500 bp 的小片段,Quast[14]软件对组装序列进行评价,通过多次优化,得到6 株大肠杆菌的基因组组装序列。应用Prokka[15]软件对组装的基因组序列进行注释,得到编码基因和非编码RNAs,使用PhiSpy[16]软件预测组装基因组中的前噬菌体区域,通过在线网 站CRISPRFinder(https://crispr.i2bc.paris‑saclay.fr/Server/)分析组装基因组中的CRISPR 序列。
1.2.3 进化类型分析 参照文献[17]合成chuA、yjaA和TspE4.C2基因的特异性引物(上海生工)。采用煮沸法提取受试菌株的基因组DNA 为模板,进行三重PCR 扩增。PCR 反应体系(20 μL):2×TaqPCR MasterMix 10 μL;上、下游引物(10 μmol/L)各0.5μL;DNA 模板1 μL;ddH2O 为6 μL。PCR 反应条件:94℃ 5 min;94℃ 45 s,退火60℃ 30 s,72℃18 s,30 次循环;72℃ 5 min;4℃保存。取3 μL 扩增产物经1.5%琼脂糖凝胶电泳检测,据检测结果鉴定大肠杆菌的系统进化群。用Abricate 软件将基因组组装序列比对到数据库ECOH[18](https://github.com/katholt/srst2/tree/master/data),据基因序列相似性鉴定菌株的血清型。利用mlst[19]软件对组装的基因组实施多位点序列分型(multilocus sequence typing,MLST)。
1.2.4 毒力因子基因的筛选与验证 将测序菌株的基因集,应用Abricate 软件与VFDB[20]数据库(http://www.mgc.ac.cn/VFs/main.htm)的核心序列集进行比对(BLAST 参数:e‑value 10-5,最小覆盖度80%,最小相似度80%),得到毒力因子候选基因。从基因组注释文件中提取各菌株分泌系统基因簇,绘制分泌系统的基因簇分布图,比较不同菌株各类分泌系统的异同。根据基因组与毒力因子数据库的比对结果,从每种毒力因子基因类型中挑选代表性的毒力因子基因(共23 个)进行PCR 验证(表1),基因组DNA 提取、PCR 反应和琼脂糖凝胶电泳同1.2.3。

表1 毒力因子基因检测引物Table 1 Primers for the detection of virulence factor genes
1.2.5 具有高影响变异的毒力因子基因的筛选与验证 将质控后各菌株的测序数据通过软件BWA 比对到参考基因组E. coli. str.K‑12 上,应用SAMtools和GATK 软件检测菌株基因组中的单核苷酸多态性(SNP)和短序列插入缺失突变(InDel),以SnpEff[21]软件对SNPs 和InDels 影响的基因进行注释,从中筛选出具有高影响变异的毒力因子基因。在毒力因子基因保守区域设计引物进行PCR 扩增(表1)。扩增产物进行双向测序(上海生工),用DNAMAN[22]软件对基因序列进行拼接,根据测序图谱校正拼接序列,将各基因的拼接序列与参考基因组进行比对,分析基因中发生的碱基变异。
2 结果
2.1 基因组序列组装与注释
经拼接(表2),毒力不同的6 株大肠杆菌最终获得108-191 条scaffolds;N50 为91-161 kb,确保了基因组组装的质量。菌株的基因组全长为4.62-5.3 Mb,对参考基因的覆盖率约为90%,其中菌株S10670 对参考基因组的覆盖率最低(87.45%),P32的覆盖率最高(91.98%)。
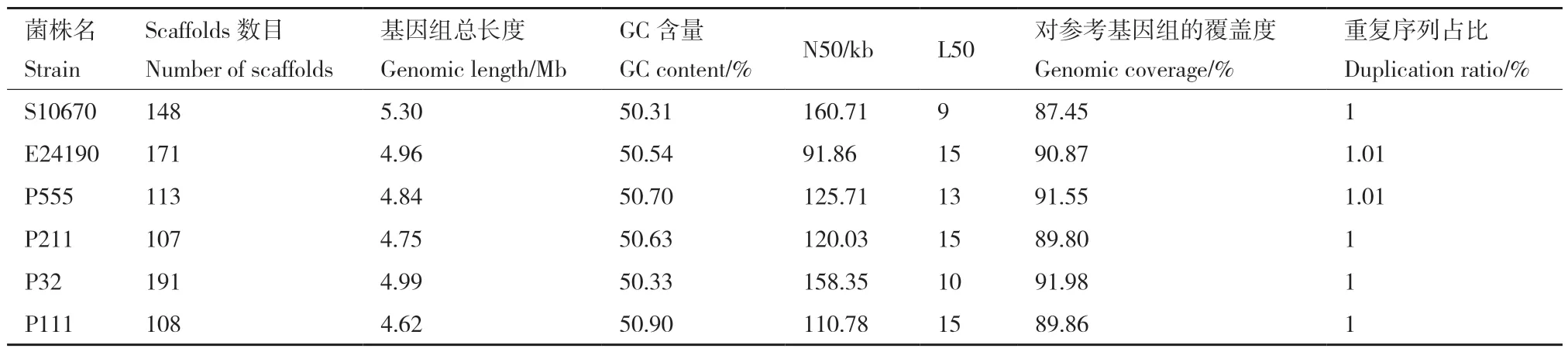
表2 菌株基因组组装质量分析Table 2 Quality analysis of the assembly genomes in E. coli strains
经Prokka 注释(表3),从6 株大肠杆菌中鉴定出4 308-5 040 个CDS 和3 364-3 557 个编码基因,普遍高于参考基因组的数量。参考菌株有88个tRNA 编码基因,检测菌株的tRNA 基因数目为83-98 个,预测到菌株的rRNA 基因显著少于参考菌株。菌株S10670 的前噬菌体数目最多(3 个),菌株E24190 和P555 基因组中没有预测到前噬菌体序列。P211 的CRISPR 序列最多,其余菌株基因组中可能存在的CRISPR 序列数量高于参考菌株。
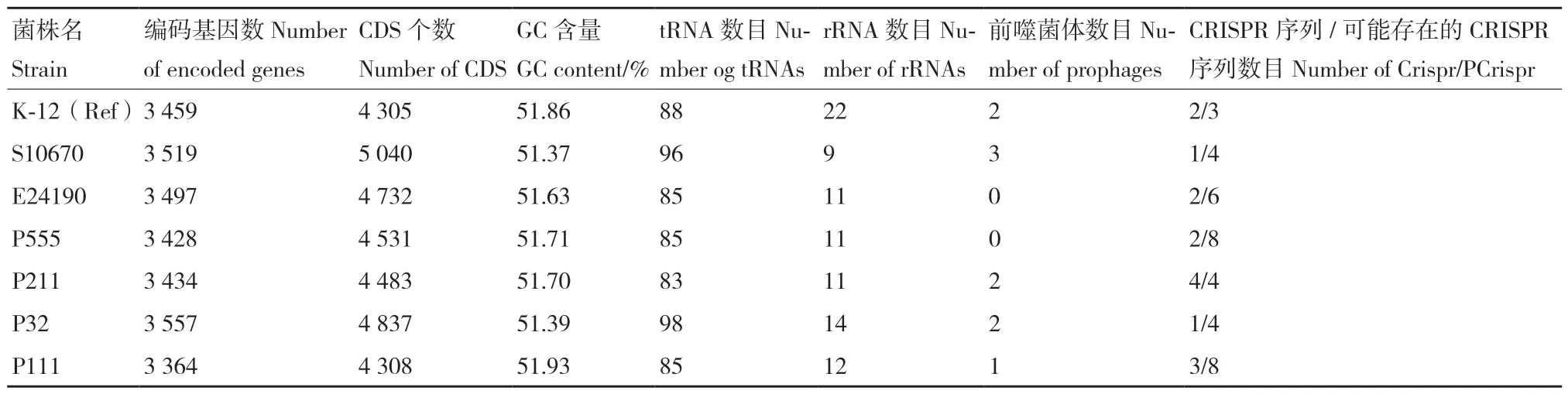
表3 基因组注释分析Table 3 Genome annotation
2.2 进化类型分析
通过三重PCR 检测和序列相似性分析,菌株S10670 属于系统进化D 群,其余菌株属于A 群。各菌株的血清型不同(表4),基于7 个管家基因的等位基因序列比对得到各菌株的多位点序列类型(sequence typing, ST)和克隆复合体类型(clonal complex, Cplx),E24190、P555、P32 为ST10 Cplx,P211 是一种新的ST 型。
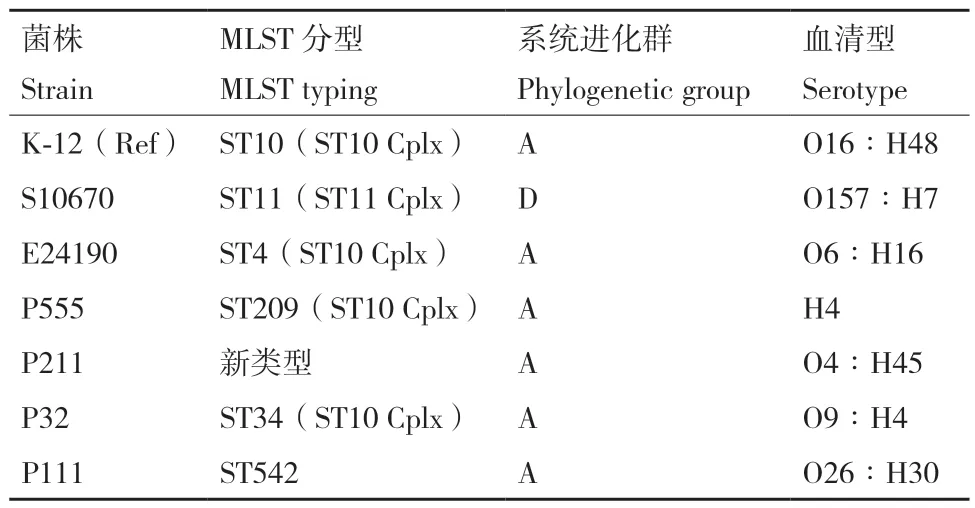
表4 系统进化群、血清型及MLST 分型Table 4 Phylogenetic groups, serotypes and MLST typing
2.3 毒力因子基因的筛选与验证
6 株大肠杆菌的毒力因子基因相比(表5),菌株之间毒力因子基因数量为112-280 个,基因数量相差1 倍以上,强毒菌株的毒素基因数量明显多于中等毒菌株和弱毒菌株。除强毒菌株S10670 外,其余菌株的鞭毛相关基因占总毒力因子基因的一半以上。菌株P211 的铁摄取和生物膜相关基因较多。各菌株的分泌系统基因数量差异较大,弱毒菌株也可以拥有丰富的分泌系统基因。
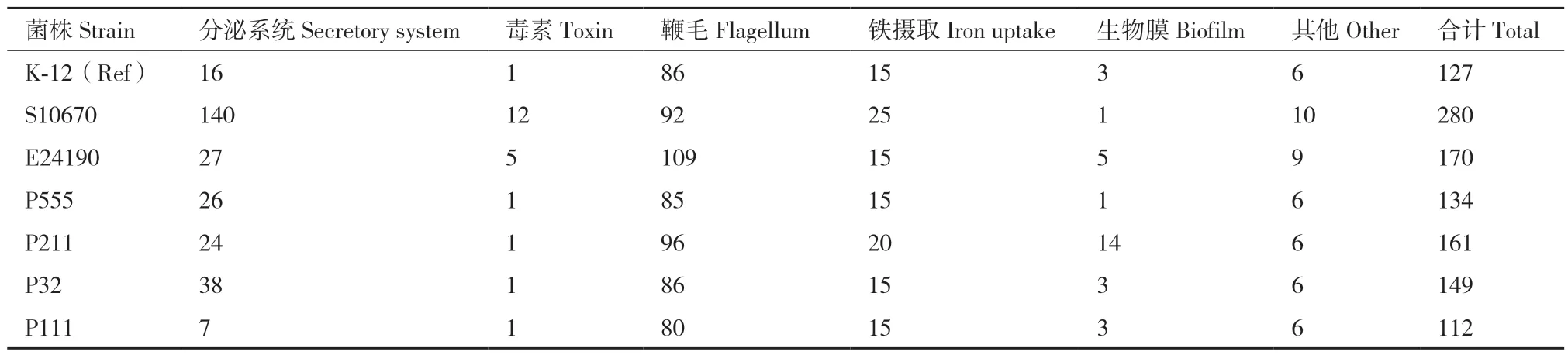
表5 菌株基因组中毒力因子分析Table 5 Virulence factors in genomes of strains
6 株致病菌分泌系统的差异集中于T2SS 和T6SS(图1)。强毒菌株中预测出全部的T2SS 基因簇(11-13 个基因),参考基因组和弱毒菌株P32 中的T2SS基因簇不完整,2 个中等毒菌株中未检测到T2SS 基因。大肠杆菌三型分泌系统(E. colitype III secretion systems, ETT)分为ETT1 和ETT2,其中ETT1 型只在强毒菌株S10670 基因组中分布;菌株S10670、P32 和P211 含有ETT2 基因,其中富含假基因,其余菌株不含ETT2 基因。菌株S10670 和P555 中预测出全部的T6SS 基因簇(19-20 个基因),E24190和P32 的T6SS 基因较少,只有3-6 个,其余菌株未检测到T6SS 基因。
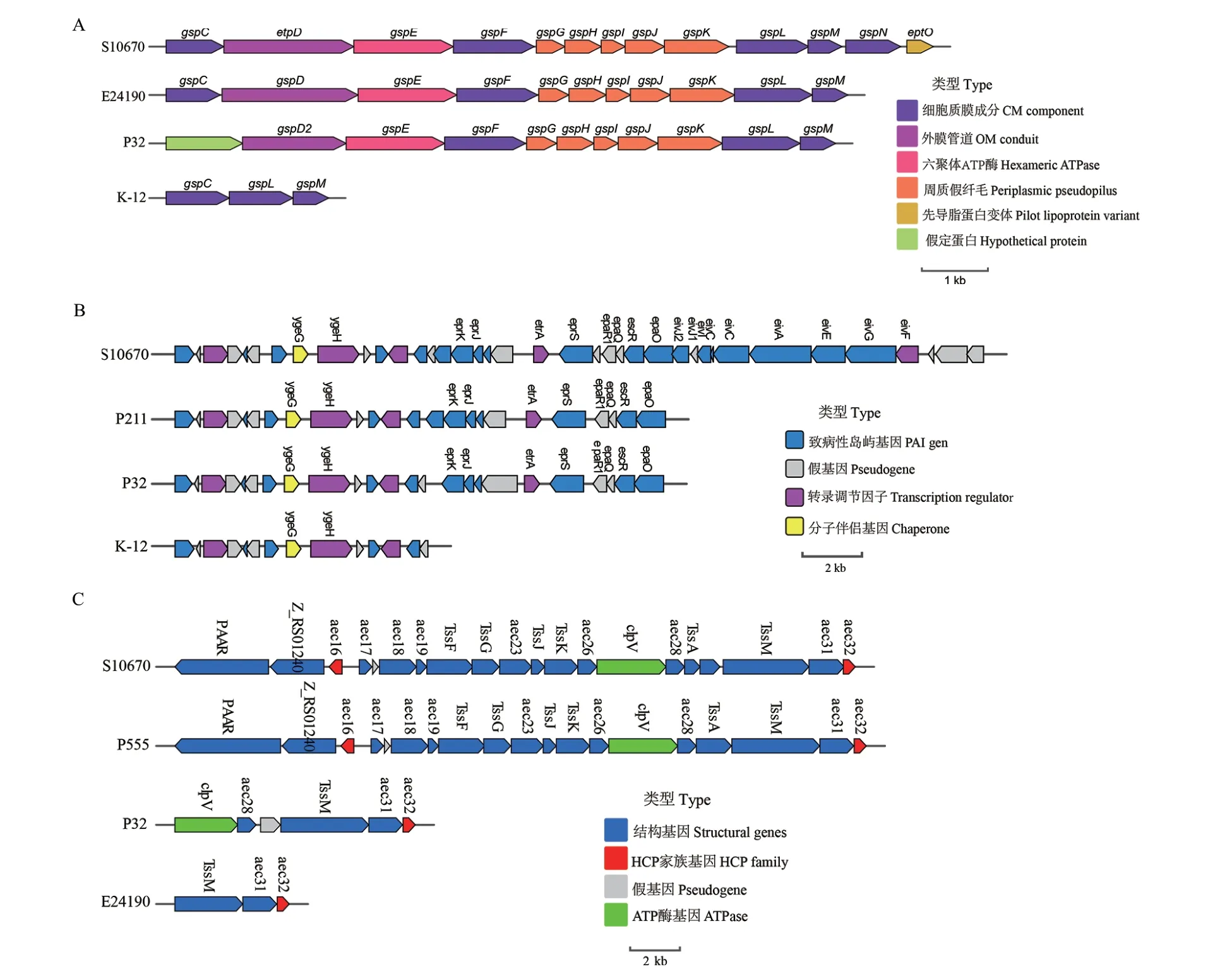
图1 六株菌的分泌系统基因簇比较Fig. 1 Comparison of the secretion system gene cluster from six strains
从各类毒力因子基因中挑选代表性的23 个毒力因子基因,采用PCR 技术进行验证(图2,表6)。从强毒菌株S10670 基因组中检测到T2SS 的外膜管道蛋白基因etpD、ETT1 的紧密黏附素基因eae、ETT2 中的转录因子eivF和脂蛋白前体基因eprK、T3SS 的效应物espX1和espO1‑2基因、T6SS 溶血素共溶蛋白家族基因aec16、血红素吸收系统基因chuA、T5SS 的ehaA和ehaB基因、志贺样毒素基因stx2A和stxB、侵袭素EaeH。强毒菌株E24190 基因组中检测到T2SS 的GspD基因和六聚体ATP 酶基因GspE,其中GspD与etpD基因同源;含有T3SS 的效应物基因espX1,T6SS 的溶血素共溶蛋白家族基因hcp1、自分泌黏附素基因aatB、耐热肠毒素和不耐热肠毒素基因、肠聚集黏附性耐热毒素基因east1,溶血素基因hlyA、侵袭素基因EaeH。

图2 23 个毒力因子基因PCR 扩增分析Fig. 2 PCR amplification of 23 virulence factor genes
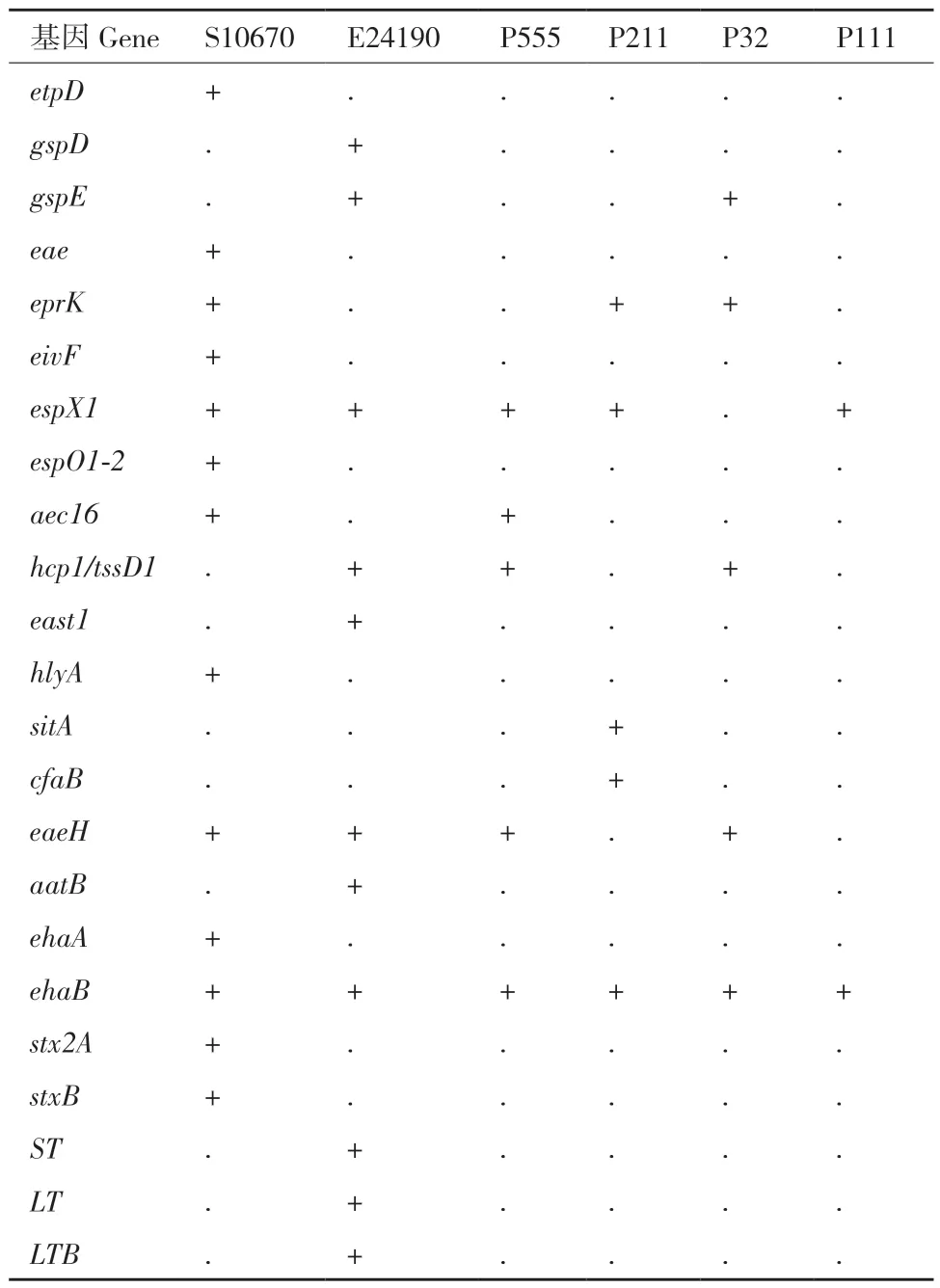
表6 23 个毒力因子基因验证汇总分析Table 6 Summary of 23 validated virulence factor genes
中等毒菌株P555 基因组中检测出espX1、aec16、hcp1、EaeH和ehaB基因;P211 中含有eprK、espX1、铁载体内膜转运系统基因sitA、ehaB基因和CFA/I 菌毛基因cfaB。弱毒菌株P32 基因组中含有GspE、eprK、hcp1、EaeH和ehaB基因;而P111基因组中只有espX1和ehaB两个毒力因子基因。上述PCR 检测结果与基因组测序和注释分析的毒力因子基因一致。
2.4 毒力因子基因变异分析与验证
对6 株致病性大肠杆菌的基因组进行SNP 和InDel 变异分析(表7),从中检测到丰富的SNP 位点,菌株S10670 拥有的SNP 最多。除菌株P111 外,其他菌株基因组中的缺失突变比插入突变数量多。进一步分析变异对基因的影响,得出受变异影响大的毒力因子基因只有7 个,分别是T3SS 效应因子基因espY1、espL4和espX4,Fim 鞭毛基因FimD和FimI,转录因子基因rpoS和尿囊素酶基因allB。采用PCR 和Sanger 测序(表8),证实菌株S10670 和P555 的espY1基因发生了一个移码突变(图3‑A);菌株E24190、P211、P32、P111 的espL4基因都缺失了612 bp(图3‑B),菌株P111 的espL4基因中还有一段超过900 bp 的插入序列,菌株P555 的espL4基因中检测到一段不同于参考基因的序列;菌株P555 的espX4基因也检测到一段超过900 bp 插入序列,S10670 的espX4基因还有两个移码突变(图3‑C);E24190 中FimD基因检测到一个无义突变(图3‑D);菌株P32 的FimI有一个移码突变(图3‑E);菌株S10670 中allB基因含有一个无义突变(图3‑F)。E24190 的rpoS基因发生771 bp 的插入变异。这些变异将导致相关基因编码的蛋白失活。

图3 毒力因子基因的高影响变异位点Fig. 3 High-impact variant loci in virulence factor genes
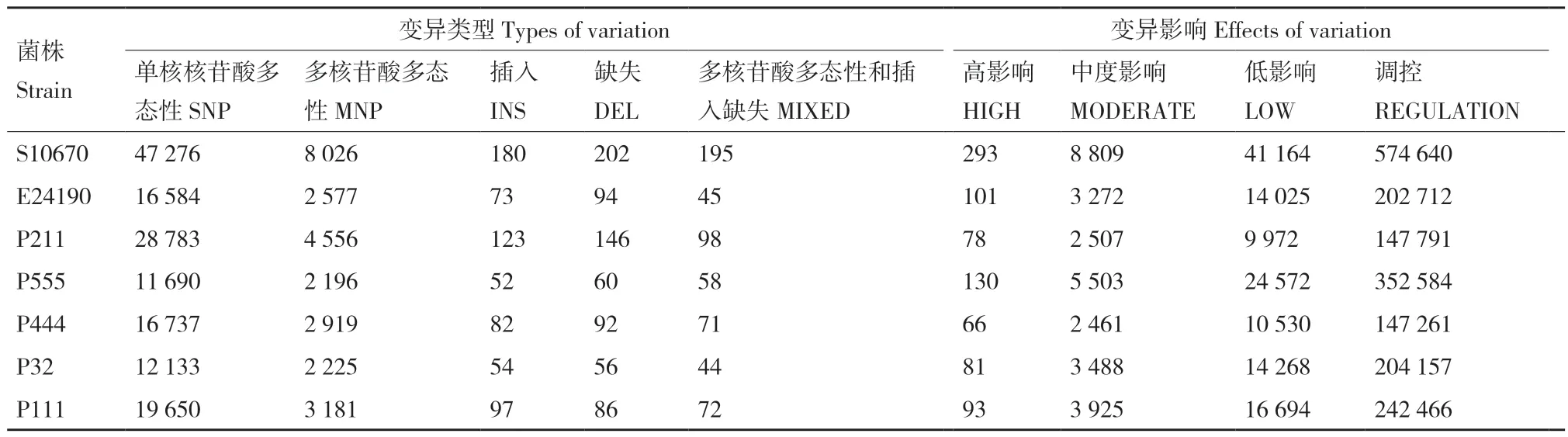
表7 各菌株的基因组变异位点Table 7 Variation sites in the genome of each strain

表8 七个具有高影响变异的毒力因子基因Table 8 Seven virulence factor genes with high impact variants
3 讨论
猪源致病性大肠杆菌是一种常见的传染性病原体,可引起严重腹泻等疾病[23]。为了有效地预防和控制猪大肠杆菌病,本研究通过基因组测序、组装,得到6 株不同毒力的猪源致病性大肠杆菌的基因组信息。相比之下,强毒菌株S10670 和E24190 的基因组较大(5.3 Mb 和4.96 Mb),基因组最小的是弱毒菌株P111,仅有4.62 Mb,与参考基因组的大小相似。参考基因组E. coli. str.K‑12 属于非致病的肠道共生菌,全长4.64 Mb,有4 288 个编码基因,与致病性大肠杆菌(O157:H7,菌株EDL933)相比少了1 387 个基因[24]。菌株S10670、E24190、P555、P211、P32、P111 的CDS 数目依次为5 040、4 732、4 531、4 483、4 837、4 308,编码基因从3 519 个降至3 364 个。总体上,随着菌株的毒力从高到低变化,基因组长度、CDS 和编码基因数量表现出逐渐降低的趋势。
大肠杆菌的致病力与种系发育分群有一定关系,B2 和D 群是主要的肠道外致病性大肠杆菌,A和B1 群多为肠道共生型大肠杆菌,四川地区猪源致病性大肠杆菌多属于A 和B1 群[25],新疆地区的多为B1 和A 群[26],本研究检测的菌株中,本地分离的4 株菌均属于A 群,提示养猪业应关注肠道共生型大肠杆菌对大肠杆菌病的影响。大肠杆菌的某些特殊血清型具有致病性[27]。本研究中,从患病猪粪样中分离的菌株血清型为H4、O4: H45、O9: H4、O26: H30,从云南分离得到101 株猪源大肠杆菌中优势血清型为O4[28],国内山东地区检测出血清型O9[29],本研究检测菌株的血清型有一定的多样性。此外,本研究检测到3 个菌株属于ST10 克隆复合群,新发现一种ST 型。山东360 株猪源大肠杆菌中,优势的复合群为ST10 克隆复合群[30]。表明ST10 克隆复合群可能是猪源大肠杆菌的优势ST 型。
大肠杆菌携带的毒力因子是决定其致病性的关键因素[31]。在感染初期,黏附素可介导细菌定植于宿主的肠道上皮细胞[32],不同致病性大肠杆菌的菌毛类型不同。本研究的强毒菌株与弱毒菌株相比拥有更多的黏附系统基因,强毒菌株拥有毒素相关的菌毛黏附素和一些毒性较强的非菌毛黏附素,以协助大肠杆菌入侵宿主细胞[33],如CFA/I 菌毛基因仅在P211 基因组中存在。
细菌的铁吸收和转运系统能帮助细菌从宿主获取铁元素,本研究检测的6 株大肠杆菌中,均含有铁吸收和转运系统中的肠杆菌素基因,铁载体内膜转运系统仅在P211 基因组中存在,血红素吸收系统仅从S10670 中检测到。构建血红素吸收系统基因chuT基因缺失突变株,证明细菌的致病性不受chuT基因的影响[34]。推测铁吸收和转运系统可能不是大肠杆菌毒力的决定性因素。
致病性大肠杆菌的外毒素主要包括志贺毒素(Stx)和肠毒素等。本实验室前期从贵州本地猪场的患病仔猪中分离到志贺样毒素和肠毒素基因[35-36]。本研究中仅在S10670 基因组中检测到志贺样毒素基因,E24190 中含有耐热肠毒素、不耐热肠毒素基因以及肠聚集黏附性耐热毒素east1基因。此外,细菌释放的溶血素(hly)可进入宿主的血液循环,在靶细胞膜上形成孔道,使大部分哺乳动物的红细胞和免疫细胞破裂[37],本研究仅从菌株S10670 基因组中检测到溶血素基因hlyA,中等毒菌株和弱毒菌株中都没有检测到溶血素基因。大肠杆菌通过志贺样毒素、溶血素等改变宿主肠道的完整性,进而引发炎症等,最终导致猪的腹泻[38]。本研究结果提示强毒力菌株可能通过多种毒素的综合作用,破坏宿主的肠道屏障致病。弱毒菌株含有的毒素基因较少,致病力较弱。
细菌利用不同的分泌系统可以将蛋白运送到细菌外。迄今从革兰氏阴性菌中发现了6 种分泌系统(即I-VI 型)[39]。T1SS 主要运输抗生素和毒素等小分子[40],与ABC 转运蛋白非常相似[41],溶血素和自分泌黏附素均属于T1SS,自分泌黏附素是由aatPABCD基因簇编码的外膜蛋白,能促进致病性大肠杆菌对宿主细胞的黏附,破坏宿主的自我保护屏障[42]。从107 株鸭源致病性大肠杆菌中,检测出28.94%的菌株含有aatA基因[43]。本研究中这两种T1SS 基因只存在于强毒菌株中。T2SS 不仅具有丰富的蛋白酶分泌活性,还能介导多种毒素向细菌外传递[44]。本研究发现强毒菌株中的T2SS 基因数量多,弱毒菌株的T2SS 基因较少。T3SS 可以将细菌的物质注入宿主细胞内,干扰宿主的免疫功能。大肠杆菌有两种T3SS,分为ETT1 和ETT2,ETT1 由LEE毒力岛编码,拥有LEE 毒力岛的致病性大肠杆菌可损伤动物的肠黏膜细胞。ETT2 在肠致病性大肠杆菌的侵袭、胞内存活和毒力等方面发挥重要作用[45],如ETT2 结构基因epaPQR簇参与致病性大肠杆菌的鞭毛形成并调控相关基因的转录,进而影响细菌的定殖能力[46]。本研究从强毒菌株S10670 基因组中检测到几乎全部的T3SS 基因,中等毒菌株P211 和弱毒菌株P32 中的ETT2 基因减少,其余菌株中未发现T3SS 基因。大肠杆菌的T5SS 是一个自主转运系统,主要由膜外功能区和跨膜区、信号肽及自分泌重复序列3 部分组成[47],主要与黏附和生物被膜的形成有关。本研究中所有菌株都检测到T5SS 的ehaB基因,但基因ehaA仅在S10670 基因组中分布。T6SS 与细菌的致病性密切相关,25%的革兰氏阴性菌拥有T6SS 基因,其中大多数为致病菌[48]。本研究中只有菌株S10670 和P555 检测到丰富的T6SS 基因20 个和19 个,菌株E24190、P32 中的T6SS 基因较少,仅有6 个和3 个。
4 结论
猪源致病性大肠杆菌基因组长度呈多态性,强毒菌株的基因组长度、CDS 和编码基因的数量高于弱毒菌株,并且拥有更多的毒力因子基因,尤其毒素基因;且强毒菌株的毒力因子基因具有较多的高影响变异。结果表明,猪源致病性大肠杆菌毒力的强弱与菌株之间毒力因子基因的数量和基因结构相关。
猜你喜欢
今日农业(2022年14期)2022-09-15
云南化工(2021年6期)2021-12-21
猪业科学(2021年3期)2021-05-21
农药科学与管理(2019年6期)2019-11-23
植物保护(2019年2期)2019-07-23
动物医学进展(2017年8期)2017-10-11
湖南畜牧兽医(2016年3期)2016-06-05
华南农业大学学报(2015年5期)2015-12-04
现代检验医学杂志(2014年5期)2014-02-02
食品科学(2013年8期)2013-03-11
