
LC-MS/MS法同时测定血浆中氟马替尼及其代谢产物浓度
2024-01-02 11:36徐月华钱洲逸赵杨黄琼叶孙鲁宁王永庆孙志明唐雯雯
医药导报 2023年12期
徐月华,钱洲逸,赵杨,黄琼叶,孙鲁宁,王永庆,孙志明,唐雯雯
(1.海安市人民医院药剂科,海安 226600;2.南京医科大学第一附属医院药学部,南京 210029;3.国家卫生健康委计划生育药品不良反应监测中心/江苏省卫生健康发展研究中心,南京 210029)
慢性粒细胞白血病(chronic myelogeneous leukemia,CML)主要发病机制为特定染色体易位(染色体9,22)形成费城染色体,导致BCR-ABL融合基因激活并表达BCR-ABL蛋白[1-2]。国内目前大多数CML患者选择伊马替尼作为一线治疗药物,并取得较好疗效[3]。氟马替尼(HH-GV678)是伊马替尼的结构类似物且在伊马替尼的基础上优化了结构,已被证明作为BCR-ABL抑制剂的效力略高于伊马替尼[4-5],并且氟马替尼能有效地克服某些KIT突变导致的耐药性[6]。与此同时,相比于伊马替尼组,腹泻和丙氨酸氨基转移酶升高等非血液学不良反应在氟马替尼组更常见[5,7]。
氟马替尼在血浆中主要以原形药物形式存在,此外存在的主要代谢物形式为 N-去甲基化代谢物M1和酰胺键水解代谢物M3,代谢物M1的稳态血浆暴露量约为原形药物的20%且氟马替尼代谢物M1与氟马替尼具有相似的药理活性[8],代谢物M3的稳态血浆暴露量约为原形药物的10%。一项对甲磺酸氟马替尼剂量相关性药动学研究表明氟马替尼的暴露量大约以剂量比例的方式增加[9]。其次氟马替尼的血浆暴露受到饮食习惯的影响[10]。为了充分研究其药代动力学药效学以便于优化治疗药物方案,同时测定、患者血浆中氟马替尼、M1和M3的浓度是必要的。本研究建立了一种液相色谱-串联质谱(liquid chromatography tandem mass spectrometry,LC-MS/MS)同时测定CML患者血浆中氟马替尼、M1和M3的方法。
1 仪器与试剂
1.1主要仪器与设备 1290 Infinity高效液相色谱仪,包括:柱温箱(G1316C)、多孔板自动进样器(G4226A)和二元高压泵(G4220A)(安捷伦科技有限公司);API 4000(美国应用生物公司);色谱工作站:Analyst®软件:1.6.3版本(美国应用生物公司);BP 211D型电子天平(德国赛多利斯公司,感量:0.01 mg);PCB-11型涡旋混合仪(德国艾本德公司);STRATOS高速冷冻离心机(赛默飞世尔科技公司);Milli-Q Gradient纯水仪(Millipore中国有限公司)。
1.2主要材料与试剂 氟马替尼(批号:505489RS-210501,纯度:95.3%,Toronto Research Chemicals公司),N-去甲基氟马替尼(M1)(批号:20210417,纯度:98%,豪森药业),氟马替尼酰胺水解产物(M3)(批号:20210330,纯度:100%,豪森药业),伊马替尼-d8(批号:2-AJK-132-1,纯度:98.7%,Toronto Research Chemicals公司),甲醇和乙腈(色谱纯,默克化工技术有限公司),甲酸(色谱纯,上海阿拉丁生化科技股份有限公司),甲酸铵(分析纯,国药化学试剂有限公司)。
2 方法与结果
2.1色谱条件 色谱柱:ACQUITY UPLC HSS T3色谱柱(2.1 mm×50 mm,1.8 μm),柱温设为38 ℃,流动相:10 mmol·L-1甲酸铵(含0.1%甲酸)(A)-乙腈(B),梯度洗脱,流速为0.5 mL·min-1,洗脱时间为6 min。洗脱程序为0 min:77% A;0~3.2 min:77%A;3.2~3.4 min:77%A→5%A;3.4~4.4 min:5%A;4.4~4.6 min:5%A→77%A;4.6~6 min:77%A。进样量为5 μL。
2.2质谱条件 采用电喷雾离子源(electron spray ionization,ESI),在正离子模式条件下,应用多重反应监测(multiple reaction monitoring,MRM)模式对氟马替尼及其代谢物进行定量分析,氟马替尼,M1,M3和伊马替尼-d8的保留时间分别为:2.30,2.20,0.56,2.10 min。其离子对、去簇电压、碰撞能量、碰撞室入口电压、碰撞室出口电压见表1。
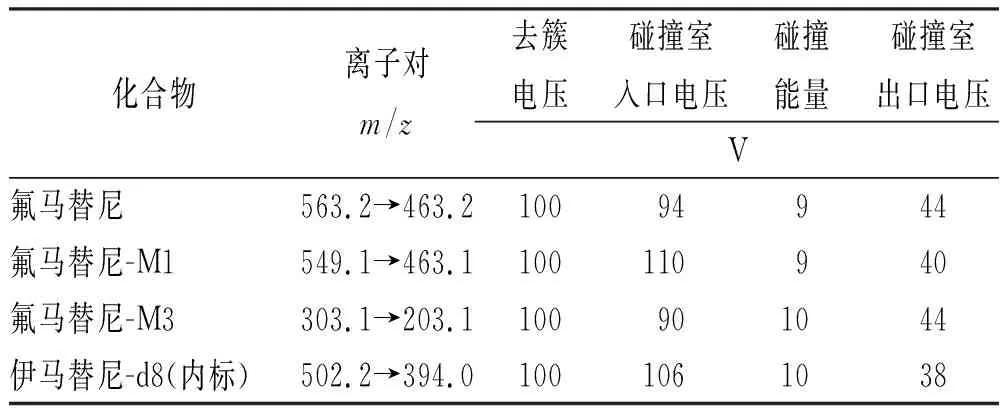
表1 氟马替尼及其代谢产物的定性离子对和碰撞电压Tab.1 Qualitative ion pair and collision voltage of flumatinib and its metabolites
2.3溶液制备
2.3.1对照品储备液 精密称取10.49 mg(经纯度校正为10.00 mg)的氟马替尼置于10 mL量瓶中,用乙腈溶解并定容至刻度,充分混合均匀,得到氟马替尼浓度为1.00 mg·mL-1的储备液,精密称取10.20 mg(经纯度校正为10.00 mg)的M1,置于10 mL量瓶中,用乙腈-二甲基亚砜(4:1,V/V)溶解并定容,充分混合均匀,得到M1浓度为1.00 mg·mL-1的储备液。精密称取10.00 mg(经纯度校正为10.00 mg)的M3置于10 mL容量瓶中,用乙腈-二甲亚砜(1:1,V/V)溶解并定容至刻度,充分混合均匀,得到M3浓度为1.00 mg·mL-1的储备液。标准曲线和质控用储备液分别配制。用甲醇将对照物质瓶中的0.5 mg伊马替尼-d8(内标)充分溶解,转移至10 mL量瓶中,用甲醇多次洗涤对照物质瓶,将洗涤液转移至同一容量瓶中,并定容,摇匀,配制成浓度为50 μg·mL-1的内标储备液。上述储备液均置于-40 ℃冰箱储存备用。
2.3.2标准曲线和质控工作液的配制 精密吸取氟马替尼、M1和M3对照品储备液,以DMSO稀释,配制氟马替尼及其代谢物质浓度均为0.008,0.016,0.040,0.160,0.400,1.60,4.00,7.20,8.00 μg·mL-1的标准曲线工作液。按上述方法配制氟马替尼及其代谢物浓度均为0.020,0.200,2.00,6.40 μg·mL-1的质控工作液。上述工作液均置于-40 ℃冰箱储存备用。
2.3.3内标溶液的配制 精密吸取20 μL质量浓度为50.0 μg·mL-1的伊马替尼-d8(内标)溶液和100 μL氨水于100 mL容量瓶中,用乙腈稀释并定容至刻度,即得10.0 ng·mL-1的含内标和0.1%氨水的乙腈溶液,置于4 ℃冰箱备用。
2.4血浆样本预处理 精密吸取血浆样本50 μL于1.5 mL EP管中,加入200 μL含内标和0.1%氨水的乙腈溶液,涡旋10 min,于4℃ 16 000 r·min-1离心15 min,离心半径为7 cm,取尽上清旋干约1 h,用150 μL 23%ACN复溶,涡旋10 min,离心10 min,取上清120 μL于自动进样小瓶中进行LC-MS/MS分析,进样量为5 μL。
2.5方法学确证
2.5.1特异性 精密吸取6个来源的人空白血浆样本50 μL,分别加入200 μL乙腈,按“2.4节”项下方法处理后进样,通过比较空白血浆样本中干扰物质的峰面积与定量下限样本中的氟马替尼及其代谢物和内标的峰面积来评价方法的特异性。空白血浆样本及空白血浆加氟马替尼及其代谢产物(浓度均为0.400 ng·mL-1)、内标样本色谱图以及人样中待测物色谱图如图1所示。结果表明,血浆样本中内源性物质不干扰化合物和内标的测定。

①空白血浆样本;②定量下限血浆样本;③氟马替尼血浆样本图1 氟马替尼和氟马替尼-M1及氟马替尼-M3在血浆中的色谱图 ①blank plasma samples;②plasma samples of lower limit of quantification;③flumatinib plasma samplesFig.1 Chromatograms of flumatinib,flumatinib-M1 and flumatinib-M3 in plasma
2.5.2线性关系考察 精密吸取空白血浆50 μL,加入含待测化合物的标准曲线工作液2.5 μL,涡旋混匀后,配制成氟马替尼及其代谢物浓度均为0.400、0.800、2.00、8.00、20.0、80.0、120、200、360、400 ng·mL-1的标准曲线血浆样本。同法用配制得含氟马替尼及其代谢物浓度均为1.00、10.0、100、320 ng·mL-1的质控样本。按“2.4节”下方法处理后进样。以待测化合物和内标的峰面积比值(Y)为纵坐标,待测化合物的质量浓度(X)为横坐标,进行线性拟合,加权因子为(1/X2),各化合物的标准曲线见表2。结果表明,各化合物的线性相关系数r均>0.99,血浆中3个化合物在各自的线性范围内线性关系良好。
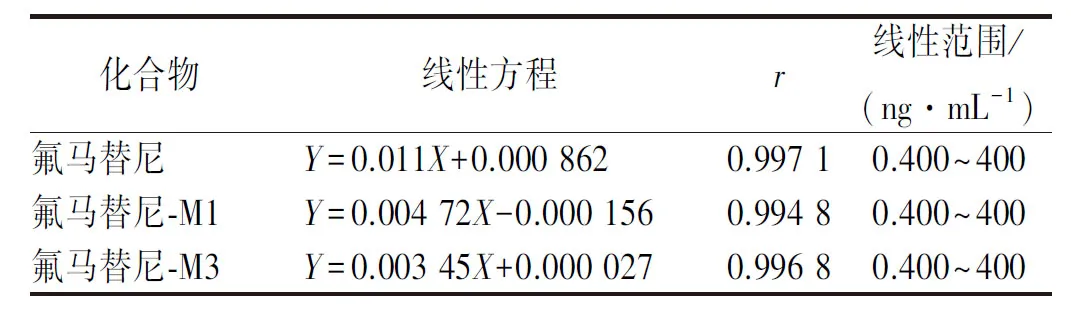
表2 线性方程、相关系数和线性范围Tab.2 Linear equations,correlation coefficients and linear ranges
2.5.3准确度与精密度 考察定量下限、低、中、次高和高5个浓度血浆质控样本精密度和准确度。定量下限及各个浓度的质控样本平行制备5份,按“2.4节”下方法处理后进行LC-MS/MS分析。日内精密度以不同浓度血浆样本同日内重复测定5次计算,日间精密度以不同浓度血浆样品3 d内重复测定3次计算,结果见表3。定量下限的批内与批间精密度均<20%,低、中、次高、高浓度质控样本的批内与批间精密度均小于15%,准确度在87.0%~111.6%。结果表明方法精密度与准确度良好。
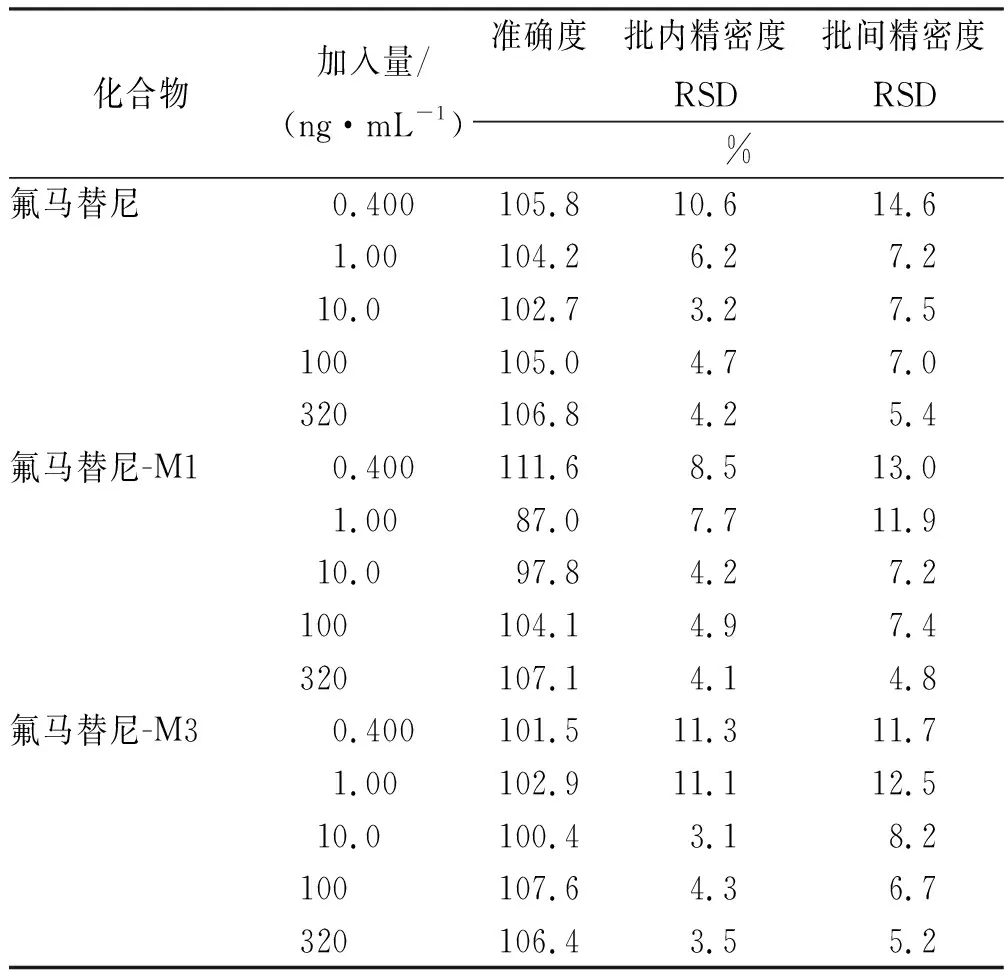
表3 氟马替尼及其代谢物化合物的准确度与精密度Tab.3 Accuracy and precision of the flumatinib and its metabolites
2.5.4基质效应和提取回收率 考察低、高质控浓度的基质效应和低、中、次高、高浓度的提取回收率。取6个来自不同个体的空白血浆基质,加入低、中、次高、高浓度质控工作液,按“2.4节”下处理后进样得到峰面积A1;另上述来源的空白血浆基质加乙腈沉淀处理后加相应质控工作液及内标配制低、中、次高、高浓度的溶液,得到峰面积A2;用纯水代替空白血浆基质,配制低、中、次高、高浓度的标准溶液后进样,得峰面积A3。基质因子=A2/A3×100%,提取回收率=A1/A2×100%。氟马替尼及其代谢物的基质因子除以内标的基质因子为归一化的基质因子。氟马替尼及其代谢的基质效应和提取回收率结果见表4。结果表明,该方法无基质效应且回收率较高。
表4 氟马替尼及其代谢物基质效应和提取回收率Tab.4 Matrix effect and recovery of flumatinib and its metabolites
2.5.5残留效应 本实验进一步考察了进样高浓度样品后的残留问题,计算进样高浓度点后的残留峰面积与标准曲线最低浓度点峰面积的比值。结果表明氟马替尼及其代谢物的残留均小于20%,残留分析物不影响检测的准确性和精密度。
2.5.6稳定性 分别制备低、高浓度的4组血浆样品,室温放置10 h、进样器72 h、长期冻存(-40 ℃和-80 ℃冻存166天)和反复冻融3次(-40 ℃和-80 ℃)条件下,按“2.4节”下方法进行测定以考察稳定性。稳定性结果见表5。结果表明,氟马替尼及其代谢物低、高浓度血浆质控样品在室温放置20 h、进样器放置72 h、3次冻融循环和-40 ℃和-80 ℃冻存166 d具有良好的稳定性。
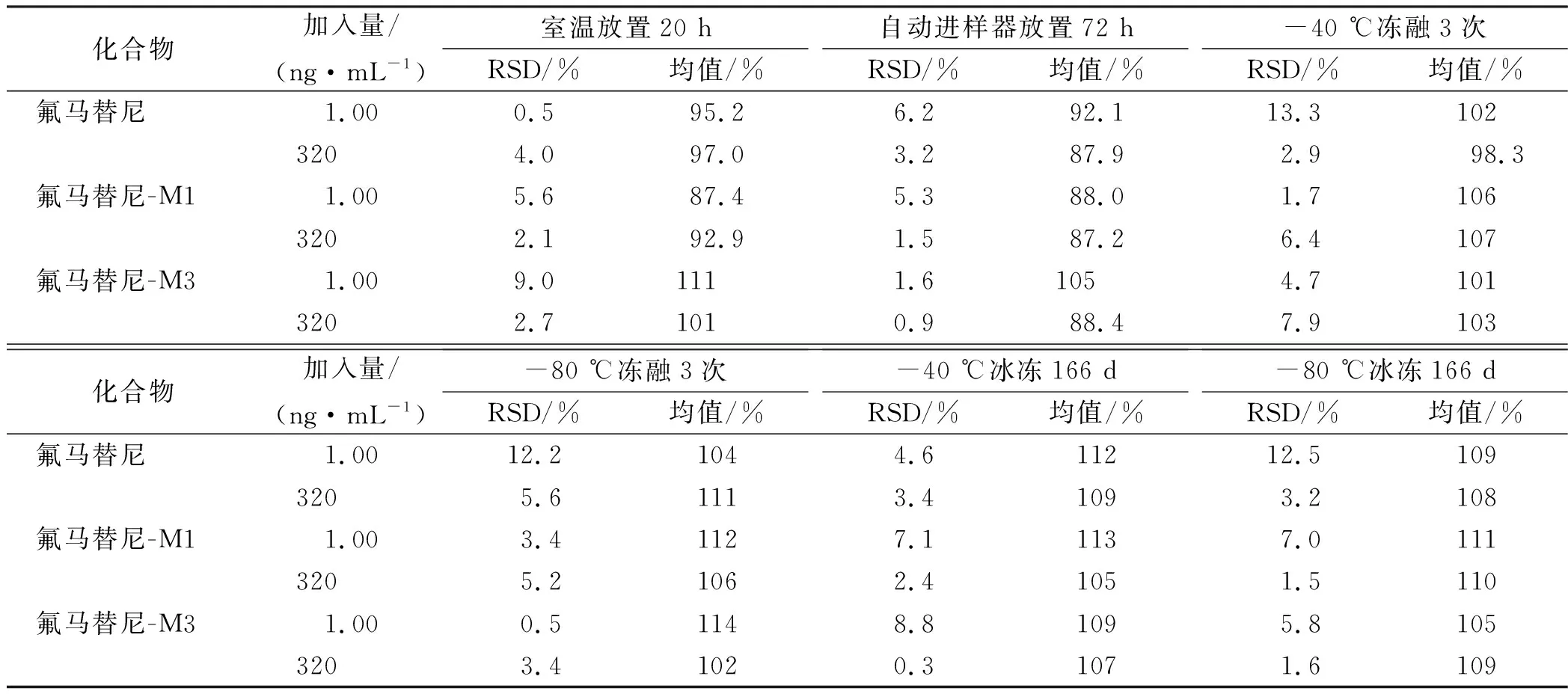
表5 氟马替尼及其代谢物稳定性Tab.5 Stability of flumatinib and its metabolites
2.6方法应用 用上述建立的方法检测本院使用氟马替尼的CML患者稳态谷浓度血浆样本。本次研究收集了本院一位非首次空腹口服甲磺酸氟马替尼片600 mg的患者服药时和服药后1.5、2、2.5、3、3.5 h的血浆样本,血样采集于含有乙二胺四醋酸抗凝管中并于-80 ℃中保存。测定的氟马替尼及其两种主要代谢物血药浓度如图2所示,根据所测结果表明 M1,M3在达峰时间2 h内表现出不明显的上升趋势,氟马替尼及其代谢物血药浓度范围内与其他药动学相关研究结果[9]一致。该患者在服药期间并没有出现非血液学不良反应如腹泻恶心等。如果需要去确定氟马替尼的血药浓度范围,则需要更多的有效性和安全性数据。

图2 1例CML患者氟马替尼及其代谢物血药浓度-时间曲线Fig.2 Blood concentration-time curves of flumatinib and its metabolites in a patient with chronic myelogeneous leukaemia
3 讨论
与YANG等[11]报道的中测定血浆中氟马替尼及其代谢物的方法相比,本方法采用梯度洗脱的方式在化合物出峰后用高有机相比例冲洗色谱柱1 min,使得基线更低,有利于减少基质效应,增强方法的重复性。本研究从质谱条件、色谱条件及血浆样品处理方法对氟马替尼及其代谢物的同时测定方法进行优化。采用ESI离子源,在正离子模式下,氟马替尼以m/z563.2→m/z370.0、M1以m/z549.1→m/z463.1、M3以m/z303.1→m/z203.1为定量离子对时响应值最高,伊马替尼-d8与氟马替尼及其代谢物在同一梯度出峰,。色谱条件以乙腈-10 mmol·L-1甲酸铵(含0.1%甲酸)为流动相,氟马替尼及其代谢物响应较好,峰型较优。前处理采用简单的蛋白沉淀法,方便,快捷,成本低,更适用于治疗药物监测,因此,本研究选择乙腈为蛋白沉淀剂。考察提取回收率时,结果显示,氟马替尼代谢物M1的回收率约30%,在沉淀剂中加入0.1%氨水后,再次考察回收率,氟马替尼及其代谢的回收率都接近100%。
本文所建立的血浆中氟马替尼及其两种主要代谢物的LC-MS/MS分析方法,操作便捷,检测限低,准确性好,能够准确定量血浆样本中氟马替尼及其两种主要代谢物的浓度,适用于临床治疗药物监测。
猜你喜欢
现代临床医学(2022年4期)2022-09-29
煤化工(2022年3期)2022-07-08
检验医学与临床(2020年1期)2020-01-10
中国资源综合利用(2016年10期)2016-01-22
分析测试学报(2015年7期)2016-01-13
应用海洋学学报(2015年2期)2015-11-22
质谱学报(2015年5期)2015-03-01
现代检验医学杂志(2015年6期)2015-02-06
食品科学(2013年15期)2013-03-11
