
Research progress on aromatization of n-alkanes
2023-11-21 12:23ZHOUQiumingWANGSenQINZhangfengDONGMeiWANGJianguoFANWeibin
燃料化学学报 2023年11期
ZHOU Qiu-ming,WANG Sen,QIN Zhang-feng,DONG Mei,WANG Jian-guo,FAN Wei-bin
(1.State Key Laboratory of Coal Conversion, Institute of Coal Chemistry, Chinese Academy of Sciences, Taiyuan 030001, China;2.University of Chinese Academy of Sciences, Beijing 100049, China)
Abstract: Conversion of saturated straight-chain alkanes generated in the deep desulfurization process of fluid catalytic cracking (FCC) gasoline and the coal-to-oil processes into aromatics via alkane aromatization is an important non-petroleum route for the preparation of aromatics that effectively improves the quality of oil.The aromatization technology of C2-C5 light hydrocarbons is relatively mature and has been used in industry.However,for the aromatization of n-alkanes,the aromatics yield is still very low due to the complex reaction process and the competition of various elemental reactions.In addition,the catalysts usually suffer from rapid deactivation.In this work,we summarize the recent advances in the aromatization of n-alkanes.The reaction mechanism of aromatization of alkanes and the effects of the dispersion of metal sites,electronic state,and acidity,morphology and pore structure of the support on the catalytic performance are discussed in detail.
Key words: n-alkane aromatization;reaction mechanism;single-(bi-) functional catalyst;metal sites;supports
Aromatics,especially light aromatics BTX(benzene,toluene and xylene),are the basic raw materials in the preparation of rubbers[1,2],plastics[3-5],adhesives[6-8]and chemical fibers[9,10],and they are also important components to improve the octane number of oil products.Nowadays,aromatics are mainly prepared through catalytic reforming process in petrochemical industry[11].The shortage of oil resources and the energy structure of “rich coal,lack of oil and gas” in China greatly limited the production of aromatics[12].
In addition,there are two major sources of gasoline in China.One is fluid catalytic cracking(FCC) gasoline,which contains high sulfur content and plenty of olefins.Although the sulfur impurity can be eliminated through deep hydrodesulfurization process,this process also leads to the conversion of olefins into saturated paraffins,thus,reducing the octane number of oil[13,14].The second is coal based catalytic synthesis oil,which yields liquid fuel from non-petroleum organic species.The Fischer-Tropsch (F-T) synthetic oil technology developed by Institute of Coal Chemistry,Chinese Academy of Sciences,has been successfully applied to several million-ton coal liquefaction industrial projects[15,16].However,this type of oil contains lots of straight chain alkanes[17,18],resulting in the decrease of octane number.Therefore,optimization and improvement of alkane aromatization technology is the key to improve the oil quality and enhance the economic benefits of coal to oil process.
At present,Cyclar[19],M2Forming[20]and Alpha techn ologies[21]have been developedforlightparaffins(C2-C5) aromatization.However,these technologies exhibit low selectivity and yield of aromatics in long chain alkanes () aromatization.Since the aromatization ofparaffins via catalytic reforming technology is mainly carried out at harsh reforming conditions,it requires the catalysts have high stability during reaction.This makes the commercial Pt supported Al2O3catalysts show low activity and selectivity in the aromatization ofn-hexane,n-heptane and otherstraight chain alkanes[22-24].Therefore,design and development of highly efficientalkanes aromatization catalyst is urgent,but challenging.
In this work,we summarize the recent research progress inn-alkanes aromatization.The reaction mechanism ofn-alkanes aromatization and the effects of metal site dispersion,electronic state,and acidity,morphology and channel structure of support on the catalytic performance are discussed.
1 Aromatization mechanism of n-alkanes
1.1 Mono-functional catalytic mechanism
The mono-functional reaction mechanism means that only a kind of active site plays crucial catalytic role during reaction.The catalyst is generally fabricated by loading metals on non-acidic support (without Brönsted acid sites),in which metal site serves as the solo active center.However,the detailed catalytic route fornalkanes aromatization over such catalysts is still controversial.The main point of contention is the order of dehydrogenation and cyclization.
As early as 1936,Kazanskii et al.[25]found that C6alkanes can be converted to aromatics on the monofunctional Pt/C catalyst.Their mechanism investigation indicated that C6alkanes were first dehydro-cyclized to six membered ring species,and then to aromatics via successive dehydrogenations.Subsequently,Davis et al.[26]employed isotope labeling method to study the reaction pathway ofn-heptane aromatization on Pt/Al2O3.14C isotope was labelled at the end and fourth carbon atoms ofn-heptane.The content of isotope labeled14C in the branch chain and benzene ring of toluene reached 80%,indicating that then-heptane aromatization was carried out through 1,6-ring cyclization pathway,as shown in Figure 1(a).Similar results were also obtained on Pt-Re and Pt-Sn bimetallic supported Al2O3catalysts[27].

Figure 1 Reaction mechanism of (a) isotope-labeled heptane to toluene[27];(b) heptatriene to toluene[28](with permission from Elsevier)
Another possible route for heptane aromatization is thatn-heptane was first dehydrogenated to heptatriene,then cyclized to cycloolefins through 1,6 or 2,7 cyclization,and finally dehydrogenated to toluene,as shown in Figure 1(b)[28].Paál et al.[29]confirmed the existence of this pathway inn-hexane aromatization at low total pressure and low hydrogen pressure.
Adolfo et al.[30]studied then-heptane aromatization mechanism on the Pt/BaKL catalyst.They found that there were two pathways during reaction.One was that the heptene generated fromnheptane dehydrogenation is first converted into methylcyclohexane through 1,6 cyclization,and then gradually dehydrogenated to aromatics.Another was that the formed heptene gradually dehydrogenated to heptatriene,which is then cyclized to aromatics.In addition,the hydrogenolysis,isomerization and other side reactions simultaneously existed in aromatization process (Figure 2).

Figure 2 Pathways of n-heptane reforming over Pt/BaKL catalyst[30](with permission from Elsevier)
1.2 Bi-functional catalytic mechanism
The bi-functional catalytic mechanism means that there are two kinds of active sites synergistically catalyzing the aromatization reaction.Generally,then-alkanes are first dehydrogenated to form active olefin intermediate species at the metal active sites,followed by the transformation of alkenes into cycloalkanes through 1,5 or 1,6 cyclization on Brönsted acid sites.Finally,the generated cyclohexane species via 1,6 cyclization can directly dehydrogenated on metal sites to produce aromatics,whereas for the cyclopentane species formed via 1,5 cyclization,it needs to first convert into cyclohexane species on Brönsted acid sites through ring expansion.
Mills et al.[31]first proposed the bi-functional catalytic mechanism on Pt/acid-treated Al2O3,in which the isomerization and cyclization mainly take place on acid sites,while metal sites are the dominant active centers for dehydrogenation.They also pointed out that conversion of alkenes to aromatics mainly follows the 1,5 cyclization plus ring expansion pathway.The reaction network was subsequently supplemented by Parera et al.[32].They indicated that cyclohexane and methyl-cyclopentane can produce corresponding alkanes through ring opening reaction on metal sites.Besides,the route of methyl-cyclopentadiene to cyclohexadiene were also proposed,as shown in Figure 3.
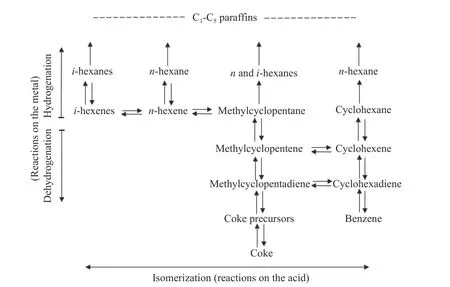
Figure 3 Bifunctional reaction scheme for reforming of C6 hydrocarbons[32](with permission from Elsevier)
Recently,Zhou et al.[33]reported two concurrent reaction pathways ofn-heptane aromatization on a highly efficient bi-functional Pt/Kβ catalyst.As depicted in Figure 4,then-heptane is first dehydrogenated to 1-and 2-heptenes on metal Pt sites,which are then cyclized to methyl-cyclohexane (MCH)and ethyl-cyclopentane (ECP) via 1,6-and 1,5-ring closure on Brönsted acid sites,respectively.The generated MCH and ECP are subsequently transformed into toluene and benzene by successive dehydrogenation,ring-expansion and dealkylation reactions.
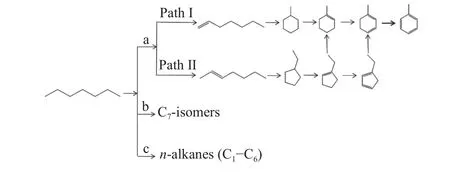
Figure 4 Reaction pathways for aromatization (a),hydroisomerization (b) and cracking (c) of n-heptane on Pt/meso-Kβ-0.05-4 catalyst[33](with permission from Elsevier)
Metal supported catalysts are widely used in the aromatization ofstraight chain alkanes.The metal site,including transition metals,alkali (earth) metals and La series metals,is the main active center of dehydrogenation.The catalyst supports can be divided into amorphous metal oxides and zeolites with specific pore structures and acidic properties,which play some roles in the cracking,dehydrogenation,oligomerization,hydrogen transfer,cyclization and isomerization.
2.1 Metal components of n-alkanes aromatization catalysts
The metal components like K,Mg,Ca,Co,Ni,Ga,Zn,Pt,Mo,etc.are widely used in aromatization reaction.In general,alkaline earth metal such as Mg and Ba can promote alkenes generation,Ga,Zn and Pt are beneficial to increase the selectivity of aromatics,while introduction of Ge,Sn,Ir,etc.can elevate the catalyst activity and stability.
The loading,dispersion degree,and states of metal sites have great influence on the catalyst performance ofn-alkanes aromatization.Smieskova et al.[34]found that decrease of the metal particle size of Zn/ZSM-5 was conducive to elevating the catalytic activity of n-hexane aromatization and the selectivity of aromatics.This is because decrease of metal particle size considerably improves the dispersion of metal sites.Wan et al.[35]compared the influence of Pd doping manner on the catalytic performance of Pt/KL innhexane aromatization.They introduced Pd into Pt/KL catalyst by impregnation (Pd-Pt/KL-IM) and hydrothermal synthesis method (Pd-Pt/KL-HD)respectively,and found that Pd-Pt/KL-IM catalyst had higher aromatics selectivity and sulfur resistance.According to the characterization results,the lower reduction temperature of Pt in Pd-Pt/KL-IM,originated from the strong electronic interaction between Pt and Pd,was benefit to improve the dehydrocyclization capacity;however,higher acidity of Pd-Pt/KL-HD catalyst resulted in serious cracking reactions.
Introduction of second metal promoter can modulate the structure and electronic state of metal sites,thus,improving the catalytic activity and stability.Hoang et al.[36]suggested that incorporation of Sn into Pt/ZrO2system prevented the catalyst deactivation and enhanced then-octane dehydrocyclization (DHC) activity.This is because the formed Pt-Sn alloy reduced the reduction temperature of Pt cluster (Figure 5(A)),thus,hindering the formation of Pt-C and suppressing coke deposition(Figure 5(B)).Similarly,the rapid aggregation of Pt was inhibited by introducing Ge into Pt/Al2O3to form Pt-Ge alloy phase,which showed higher catalytic stability and aromatics selectivity inn-octane aromatization[37].He et al.[38]systematically studied the effects of preparation methods of Pt-Zn/UZSM-5 on the catalytic performance forn-octane aromatization.They indicated that the Pt-Zn/UZSM-5-II prepared by a strong electrostatic adsorption (SEA) method displayed higher activity and aromatics selectivity than the samples synthesized by traditional co-impregnation method (Pt-Zn/UZSM-5-I),as shown in Figure 6(a).This can be rationalized that using SEA method induced the occupation of Zn atoms in the inner pore sites,thus,driving more Pt clusters to deposit on external surface of UZSM-5 support and enhancing the activity of octane dehydrogenation (Figure 6(b)).
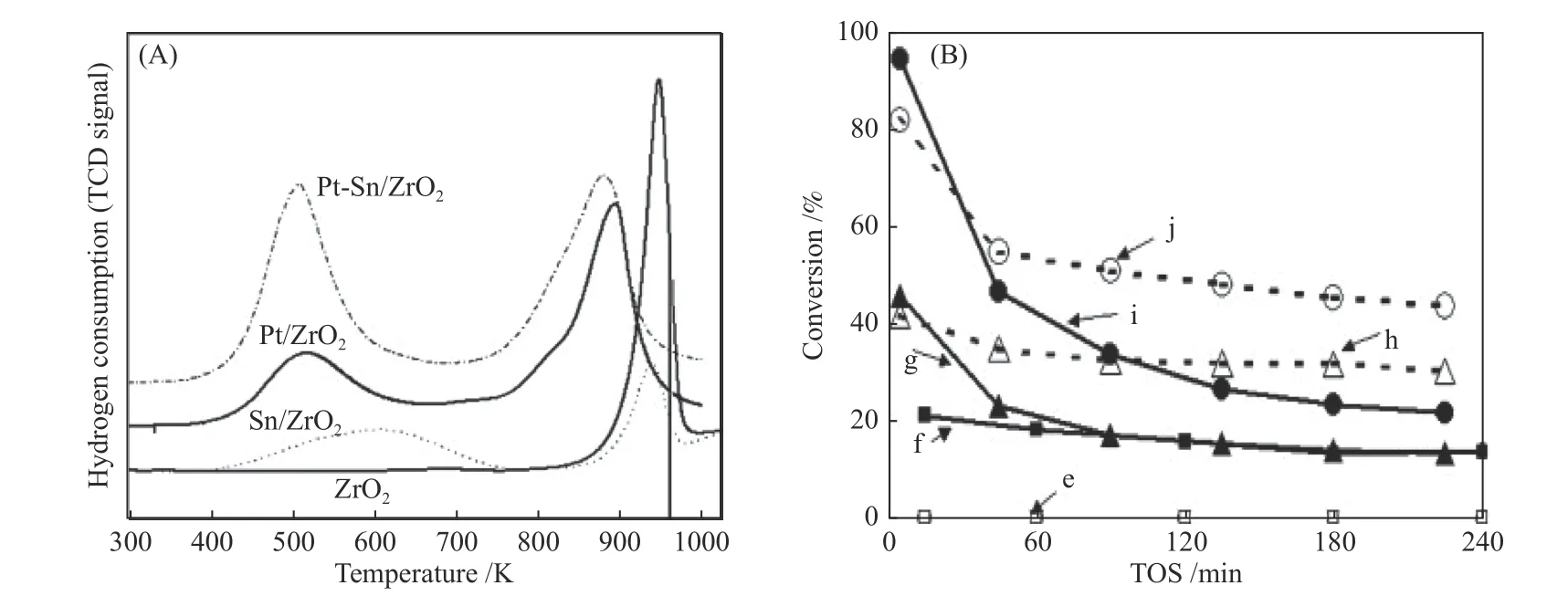
Figure 5 (A) H2-TPR profiles of virous samples;(B) n-octane conversion in n-octane dehydrocyclization (DHC) at 823 K on (e) ZrO2 in water vapor-hydrogen (WVH2) atmosphere,(f) ZrO2 in H2 atmosphere,(g) Pt/ZrO2 in H2 atmosphere,(h) Pt/ZrO2 in WVH2 atmosphere,(i) Pt-Sn/ZrO2 in H2 atmosphere and (j) Pt-Sn/ZrO2 in WVH2 atmosphere[36](with permission from Elsevier)

Figure 6 (a) Selectivity of aromatics and C6-C8 alkanes in octane conversion over various Pt/ZSM-5 catalysts; (b) Energy profiles for n-octane dissociation to form 1-C8H16 and H2 on the Pt(111) surface and Pt8 clusters distributing in the inner pores of the catalyst[38](with permission from American Chemical Society)
2.2 Supports of n-alkanes aromatization catalyst
2.2.1 Metal oxide support
Metal oxides,such as Al2O3and ZrO2,are widely used as supports in aromatization reaction due to their high mechanical strength and structural stability.Davis et al.[39,40]reported that after chlorination,the acidic Pt/Al2O3bifunctional catalyst shows higher aromatic formation rate and selectivity than those of non-acidic Pt/Al2O3monofunctional catalyst inn-octane aromatization.However,high reaction temperature easily induces the phase transformation of Al2O3that decreases the specific surface area and leads to the aggregation of loaded metal sites[41].Therefore,SiO2,TiO2,ZrO2and rare earth oxides are generally used as promoters to enhance the thermal stability of Al2O3support. For example, Pt/ZrO2-γ-Al2O3showed higher toluene yield (33.0%) than those of unmodified Pt/γ-Al2O3(24.1%). This is because the addition of Zr species can improve the stability of Al2O3support,thereby increasing the dispersion of Pt and the aromatization performance[42].
ZrO2carrier has also attracted extensive attention due to its high thermal stability, suitable acid-base and oxidation-reduction properties. It has been proved that the formed Zr3+species on ZrO2support is important Lewis acid center in aromatization process[43].Trunschke et al.[44]prepared a La2O3-modified CrOx/ZrO2catalyst, which obviously increased the aromatics yield inn-octane aromatization (Figure 7).They suggested that introduction of La2O3promoted the dispersion of free Cr3+centers and reduced the acidity of catalyst support, thus facilitating the dehydrogenation and inhibiting cracking side reaction.

Figure 7 Aromatization of n-octane over CZ (CrOx/ZrO2) and CLZ (CrOx/La2O3-ZrO2) catalyst at 823 K, W/F = 58 and TOS = 90 min[44](with permission from Elsevier)
2.2.2 Zeolite support
Zeolites with specific pore structure and tunable acidic properties have excellent catalytic performance in olefin/alkane aromatization. At present, ZSM-5 and L-type zeolites are the most widely used supports forn-alkanes aromatization.
The three-dimensional framework structure of ZSM-5 zeolite with two intersecting ten-membered ring channels results in its unique shape selective confinementeffect onaromatizationof light hydrocarbons (C2-C5)[45-47].However,metalsupported HZSM-5 catalyst (M/HZSM-5) has strong acidity,easily leading to over-cracking ofn-alkanes.Moreover, the medium-pore structure (0.54 nm × 0.56 nm for straight channel; 0.52 nm × 0.58 nm for sinusoidal channel) of ZSM-5 restricts the diffusion of long-chain alkane reactants and their corresponding aromatization products. Therefore, the aromatization performance ofstraight chain alkanes on M/HZSM-5 catalyst is not as high as expected. Further improvement of the diffusion ability and adjustment of the acidity of M/HZSM-5 are necessary.
Increasing the silicon to aluminum ratio (Si/Al) of the synthesis solution and using acid-alkali posttreatment can regulate the acidity of HZSM-5 zeolite.Sahoo et al.[48]prepared a series of dealuminated ZSM-5 catalysts by adjusting the steam treatment temperature (300-600 °C) forn-heptane aromatization.It was found that the acid content of dealuminated ZSM-5 (ZSM-5-DA) decreased with the increase of steam treatment temperature. Among them, the ZSM-5-DA prepared at 400 °C (ZSM-5-DA-400) had the highest aromatic selectivity of 35.6%.Viswanadham et al.[49]confirmed that ZSM-5-DA-400 catalyst also performed excellent aromatics selectivity inn-hexane andn-octane aromatization.In addition,introduction of mesopores and macropores into microporous ZSM-5 zeolite can reduce the diffusion resistance and enhance the catalytic stability.Li et al.[50]synthetized mesoporous HZSM-5 catalyst through KOH posttreatment method,which exhibits higher catalytic activity and aromatics selectivity than traditional microporous HZSM-5 onn-hexene aromatization.
Zhou et al.[51]prepared a Pt/KZSM-5(deAl)catalyst,which is modified by ammonium hexafluorosilicate (AHFS) combined with potassium carbonate (K2CO3) post-treatment method to controllably regulate the pore structure and acidic property of ZSM-5 zeolite support.In comparison to the unmodified Pt/HZSM-5 sample,the Pt/KZSM-5(deAl) exhibited much higher aromatics selectivity(>75%) and catalytic stability (>80 h) onn-heptane,noctane andn-nonane aromatization,as shown in Figure 8(A).The characterization results indicated that using AHFS and K2CO3modifications can generate more mesoporous and reduce strong acid sites of HZSM-5 (Figure 8(B)).This not only facilitated the diffusion of aromatic products,but also inhibited the over-cracking reaction.
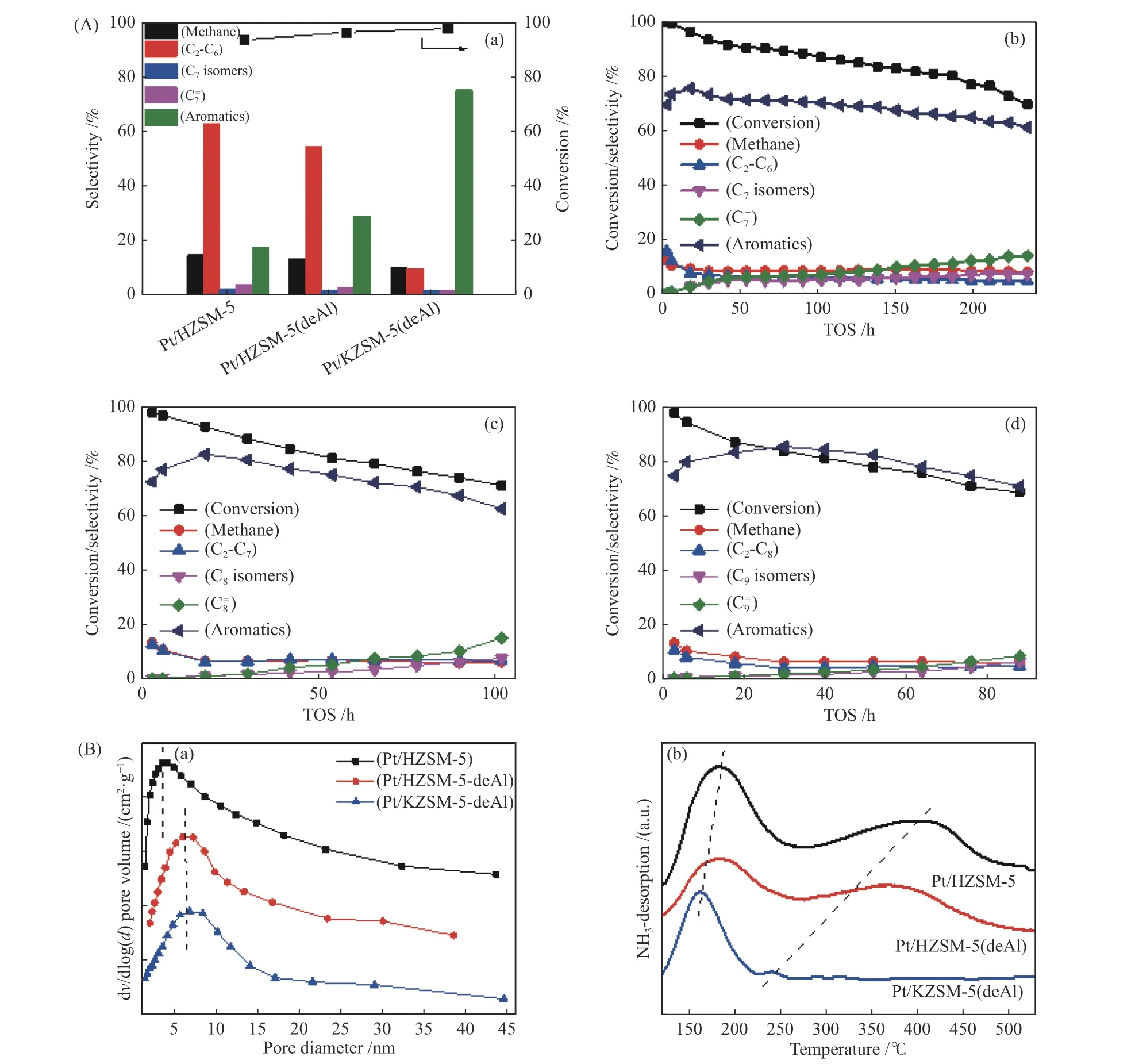
Figure 8 (A) Conversion and products distribution for n-heptane aromatization over different Pt/ZSM-5 catalysts at TOS of 12 h (a),catalytic stability of Pt/KZSM-5(deAl) catalyst for the aromatization of n-heptane (b),n-octane (c) and n-nonane (d);(B) pore size distributions (a) and NH3-TPD profiles (b) of various Pt/ZSM-5 catalysts[51](with permission from Royal Society of Chemistry)
L zeolite has one-dimensional twelve-membered ring straight channel structure with the pore diameter of 0.71 nm.Bernard et al.[52]identified that Pt supported KL(Pt/KL) catalyst had higher activity and selectivity fornhexane aromatization than that of the Pt/Al2O3.Unlike the traditional bifunctional aromatization mechanism,the Brönsted acid center is not essential for aromatics formation on Pt/KL.On the contrary,increase of the acid content of Pt/KL usually results in the decrease of aromatic yield.This may be because of the interaction between alkaline KL zeolite support and metal sites,altering the electronic properties of Pt and the catalytic performance[53].Derouane et al.[54]believed that the specific pore structure L-type zeolite can promote the formation of aromatics.Moreover,regulation of the dispersion,distribution and electronic properties of Pt active sites is regarded as effective method to enhance the catalytic performance of Pt/KL in aromatization.
The F-modified Pt/FKL catalyst was prepared for catalyzing the aromatization of C6-C7hydrocarbons[55].It was found that the aromatics yield was increased from 74.6% of unmodified Pt/KL to 81.5% of Fmodified Pt/FKL.This is because the introduced F can reduce the Pt clusters particle size and increase the dispersion of Pt,thus,promoting the dehydrogenation and aromatization activity.Xu et al.[56]fabricated a series of Pt/KL catalysts using atomic layer deposition(ALD) technique.The results suggested that Pt sites distributed in the channel of KL zeolite were more conducive to producing aromatics inn-heptane aromatization.Moreover,doping proper amount of Co on Pt/KL (Pt-Co/KL) can also promote the dispersion of Pt due to formation of Pt-Co bimetallic clusters,which increases the aromatics selectivity from 60% to 89.1% (Figure 9)[57].Although Pt/KL catalysts give higher aromatic selectivity,the poor thermal and hydrothermal stability of KL zeolite make such catalysts show low catalytic stability.Moreover,the one-dimensional straight channel structure of KL zeolite also leads to the rapid coke deposition[58-60].
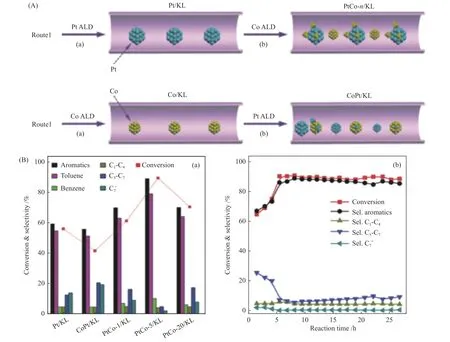
Figure 9 (A) Schematic process for the synthesis of bimetallic PtCo-n/KL (n=1,5,and 20) and CoPt/KL catalysts;(B) Catalytic performances of the catalysts: (a) Conversion of n-heptane and product selectivity over the catalysts after 5 h test and (b) Catalytic results as a function of reaction time in the aromatization of n-heptane over PtCo-5/KL catalyst[57](with permission from Royal Society of Chemistry)
Recently,then-heptane aromatization catalyzed by hierarchical Kβ zeolite was reported.Shi et al.[61,62]employed organic silane as the pore-enlarging agent to synthesize Hβ with micro-mesoporous structure (HBeta-HS),which was further ion-exchanged by different concentrations of KNO3aqueous solutions to form K-Beta-HS-x support.Upon loading Pt,the Pt/KBeta-0.2M showed excellentn-heptane aromatization performance.The catalyst lifetime was more than 200 h,and the aromatics selectivity was up to 80%(Table1).The results of acidic characterization revealed that incorporation of K+can shield most of acid sites of H-Beta-HS zeolite (Figure 10(A)), thus inhibiting the over-cracking reaction. In addition, the XPS results corroborated that K ++candonatesome electrons to Pt (Figure 10(B)),which enhances the dehydrogenation ability.The three-dimensional twelvemembered ring channel structure of β zeolite also facilitates the diffusion oflong chain hydrocarbons,thus increasing the coke resistance of catalyst.
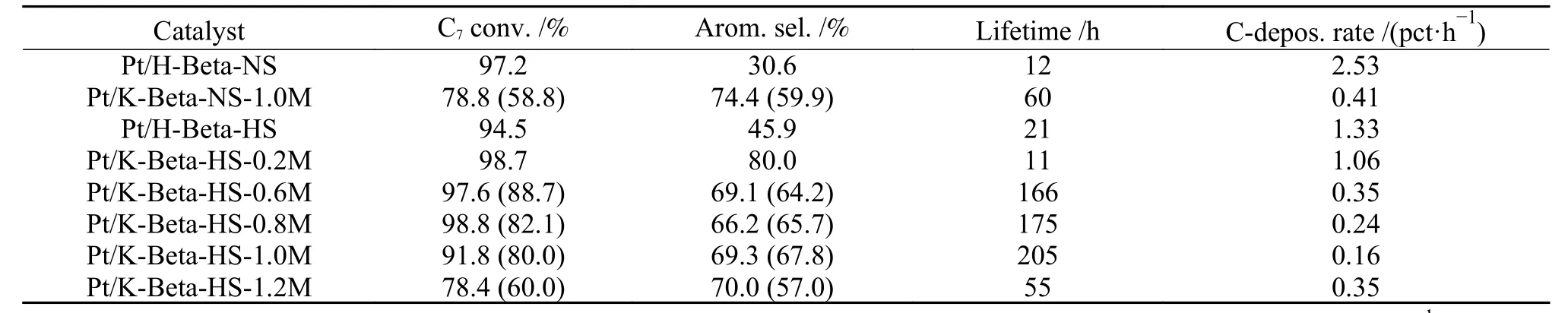
Table 1 Conversion of n-heptane,selectivity to aromatics and lifetime of various catalysts in the aromatization of n-heptane[61](with permission from Elsevier)

Figure 10 (A) NH3-TPD profiles (I) and Py-IR spectra (II);(B) Pt 4f XPS spectra of various catalysts[61](with permission from Elsevier)
Moreover,the morphology of zeolite may also have someinfluencesonthecatalytic performanceinalkanesaromatization.Zhao etal.[63]synthesized a nano-sized BaKL zeolite with particle size of around 200-300 nm by addition of Ba promoter.In contrast to the un-modified KL zeolite with larger size of 500 nm,the Pt/BaKL catalyst showed higher catalytic stability and C8aromatics selectivity inn-octane aromatization,because of its smaller particle size reducing the diffusion resistance to prevent the secondary reaction.Similarly,the hierarchical Pt/K-Beta-HS-1.0M sample also gave longer catalyst lifetime (205 h) than the traditional microporous Pt/K-Beta-NS-1.0M (60 h) inn-heptanearomatization(Table1),due toless diffusion resistanceofthe formerthanthelatter[61].
3 Conclusions
In summary,clarification of reaction mechanism and design of catalysts with high activity,high aromatic yield and long catalytic stability are the key forn-alkane aromatization process.The aromatization ofn-alkanes can be carried out through mono-functional and bi-functional pathways,which depend on the structure and composition of catalysts.For mono-functional pathway,metal site is the dominant active center for dehydrogenation and cyclization reactions.Increase of the metal dispersion,regulation of the electronic state of metal sites and introduction of secondary metal promoter are regarded as effective method to improve the resistance of metal to sintering.For bifunctional pathway,dehydrogenation is still mainly performed on metal sites,whereas for cyclization,acid sites play more vital role.Except for metal modification,regulation of acidity,morphology and pore structure of supports is also essential.Excessive acid sites of zeolite support will induce serious side reactions,such as cracking and isomerization,which decrease the aromatics yield.Therefore,it is necessary to decrease the catalyst strong acidity through altering the Si/Al ratio,and using dealumination or alkali metal ion-exchange posttreatment method.In addition,modulation of zeolite morphology and channel structure by reducing particle size and introducing hierarchical pore can effectively reduce the diffusion resistance and further elevate catalyst stability.
