
马铃薯蛋白酶抑制剂酶解产物超滤组分的抗氧化活性与结构特征
2023-11-16 11:06李苏红赵起越于思琦邹沅辛李拖平
中国粮油学报 2023年9期
李苏红, 赵起越, 于思琦, 杨 慧, 邹沅辛, 陈 晨, 李拖平
(沈阳农业大学,沈阳 110866)
马铃薯是我国第四大主食作物,其营养研究日益受到关注。但一方面由于马铃薯中蛋白质含量较少,仅占鲜质量的1.6%~2.1%,对其相关的理论与应用研究的重视不足,另一方面,作为马铃薯主要深加工产品的淀粉生产中每吨产物生产3.5 t富蛋白废液,直接排放将会造成环境污染和资源浪费[1、2]。在马铃薯全蛋白中,马铃薯蛋白酶抑制剂(PPIs)约占总蛋白质量的50%,一直被作为抗营养因子来研究,近年来其功能性如抗菌性[3]、抗氧化活性、抗肿瘤[4]等受到关注。前期的研究发现,PPIs对具有一定的抗氧化能力,但对一些自由基的清除能力较弱,如羟自由基[4]。酶水解是从蛋白质分子中释放抗氧化肽的有效方法,大分子蛋白质经蛋白酶水解后变成小分子量的肽甚至氨基酸,暴露出更多能与氧化剂反应的活性肽或者氨基酸,进而提高其抗氧化活性[5]。同时,酶解后产物的分子量变小,粒径降低有助于提高其生物可及性[6]。但是PPIs作为酶抑制剂,对某些酶的活性具有抑制作用,因此可以采用物理技术(如超声、红外照射、微波加热)进行预处理,打开蛋白质的内部结构,暴露出更多的酶切位点,有助于提高酶解效率[7]。虽然红外光照射、微波加热、超声波均是通过引入额外能量快速酶解的策略,但前两者过高功率下具有一定危险性,所以实验选择超声波[8]。
研究旨在提高PPIs 的抗氧化能力,采用超声辅助酶解法制备马铃薯蛋白酶抑制剂酶解产物(PPIHs),并对其进行超滤分离,分析PPIHs及超滤各组分的抗氧化活性和结构特性,为马铃薯蛋白资源的开发和利用提供技术参考。
1 材料与方法
1.1 材料与试剂
马铃薯、大豆油;蛋白酶K(30 U/mg);DPPH、ABTS;8-苯胺-1-萘磺酸(ANS)、水杨酸、亚油酸,化学试剂均为分析纯。
1.2 仪器与设备
CR21N高速冷冻离心机,DYCP-31DN电泳仪,VCX-750超声波细胞破碎仪,Scientz-/ZN/A冷冻干燥机,STIGE-2傅里叶变换红外光谱分析仪,3 000Zetasizer nano ZS纳米粒度及Zeta电位分析仪,F-7000荧光分光光度计。
1.3 方法
1.3.1 制备马铃薯蛋白酶抑制剂
参照Liu等[9]的方法制备马铃薯蛋白酶抑制剂(PPIs)。在马铃薯汁中加入饱和度为40%的硫酸铵,搅拌2.5 h使其充分溶解,10 000 r/min离心20 min后取沉淀用水进行复溶得到马铃薯全蛋白,将其溶液的pH调到3.0后静置0.5 h后再离心除去马铃薯糖蛋白Patatin,Patatin是一种马铃薯储藏蛋白,约占马铃薯可溶性块茎蛋白的40%,由于马铃薯品种的不同,其分子质量分布在40~45 ku范围内[10]。再将沉淀在80 ℃中加热10 min除去高分子量蛋白和剩余的Patatin,离心后取上清透析36 h,冷冻干燥。
1.3.2 超声辅助酶解马铃薯蛋白酶抑制剂
称取适量PPIs粉末溶于蒸馏水中(5 mg/mL),在超声波功率为450 W下超声30 min,在底物质量浓度为5 mg/mL,酶底比(酶与底物浓度的比值)为5%,酶解时间为2.5 h,酶解温度为58 ℃的条件下,用1 mol/L NaOH调节溶液至蛋白酶K的最适酶解pH 8.0,再将底物置于58 ℃水浴锅中预热5 min后加入质量分数5%蛋白酶K进行酶解2.5 h。酶解结束后置于沸水浴中灭活5 min,冷却后,8 000 r/min离心5 min,收集上清液并冷冻干燥成粉备用。
1.3.3 超滤分离抗氧化活性肽
将500 mg的PPIHs配成0.1 mg/mL的溶液放入不同截留分子质量的超滤离心管内进行4 000r/min离心10 min,重复4~5次,使分子量的截留值为10 ku和3 ku,得到>10 ku(PPIHs-Ⅰ)、3~10 ku(PPIHs-Ⅱ) 和<3 ku(PPIHs-Ⅲ) 3个不同分子量的组分,冷冻干燥后,其质量分别为m1、m2和m3,并放在-20 ℃保存。使用式(1)~式(3)计算各组分的得率:
PPIHs-Ⅰ的得率=(m1/500)×100%
(1)
PPIHs-Ⅱ的得率=(m2/500)×100%
(2)
PPIHs-Ⅲ的得率=(m3/500)×100%
(3)
1.3.4 Tricine-SDS-PAGE 分析
根据Liu等[9]的方法,采用质量分数15.5%分离胶和4.0%浓缩胶进行Tricine-SDS-PAGE,分析马铃薯蛋白酶抑制剂及其酶解产物的分子质量分布。
1.3.5 酶解产物的理化性质
1.3.5.1 溶解度
参照Kim等[11]方法并做修改。将PPIHs溶于蒸馏水中配制成1%(质量浓度)的溶液,用1 mol/L的NaOH或HCl调节pH,使用磁力搅拌器在室温下搅拌2 h后,以4 000 r/min离心10min。倒出上清液并通过Whatman#4滤纸。滤液用于测量总氮含量。溶解度计算见式(4)。
溶解度=(TNs/TNi)×100%
(4)
式中:TNs和TNi分别是1%的PPIHs及其滤液中氮含量。
1.3.5.2 起泡性和泡沫稳定性
参照Liu等[9]方法并加以修改。将30 mL(V0)的PPIHs(10 mg/mL)溶液在均质机以10 000 r/min的速度搅拌2 min,然后马上移到200 mL量筒里,迅速记下此时泡沫的体积为V1,静置0.5 h后,再记录一次泡沫的体积为V2,根据式(5)和式(6)分别计算起泡性和泡沫稳定性。
起泡性=(V1-V0)/V0×100%
(5)
泡沫稳定性=(V2-V0)/V1×100%
(6)
式中:V0为未处理体积;V1为处理后的体积;V2为静止后的体积。
1.3.5.3 乳化性和乳化稳定性
参照Liu等[9]的方法,并加略微调整。将不同pH的PPIHs溶液(10 mg/mL)与大豆油按照3∶1的比例用内切式匀浆机以10 000 r/min的速度搅拌2 min。利用质量分数0.1% SDS溶液,把样品溶液再稀释250倍,在500 nm处测定吸收度为A,15 min后再测1次该溶液的吸光度为A15,用0.1% SDS溶液调零。使用式(7)和式(8)分别计算乳化性和乳化稳定性:
乳化性/(m2/g)=[(2×2.303)/C×(1-ψ)×10 000]×A×N
(7)
乳化稳定性=A15/A×100%
(8)
式中:C为样品的质量浓度/g/mL;ψ为油的体积分数;N为样品稀释倍数。
1.3.6 体外抗氧化活性的测定
1.3.6.1 羟自由基的清除能力
参照Liu等[9]方法略作修改,在试管中依次加入1 mL的样品溶液、FeSO4溶液(9 mmol/L)、H2O2(8.8 mmol/L)和乙醇-水杨酸溶液(9 mmol/L),然后放在37 ℃水浴锅中反应15 min,4 500 r/min离心5 min,在510 nm测定吸光度AX。空白组用蒸馏水取代PPIHs溶液,吸光度为A0。样品对照组用蒸馏水取代H2O2,吸光度为AX0。使用式(9)计算羟自由基清除率:
羟自由基清除率=(A0+AX0-AX)/A0×100%
(9)
1.3.6.2 DPPH自由基的清除能力
参照Liu等[9]方法略作修改,分别吸取同体积的样品溶液和0.04 g/L的DPPH溶液加入试管中,混匀后马上放在25 ℃水浴锅中避光反应30 min,在4 500 r/min条件下离心10 min后放在517 nm测吸光度,为AX。空白组用无水乙醇取代PPIHs溶液,吸光度为A0。样品对照组用无水乙醇取代DPPH溶液,吸光度为AX0。其中无水乙醇为参比溶液。使用式(10)计算DPPH自由基清除率:
DPPH自由基清除率=(A0+AX0-AX)/A0×100%
(10)
1.3.6.3 ABTS自由基的清除能力
参照Liu等[9]方法略作修改,将样品溶液和 ABTS工作液按照体积比为1∶4加入试管中,反应6 min后在734 nm处测吸光度为A。空白组用95%乙醇取代PPIHs溶液,吸光度为A0。使用式(11)计算ABTS自由基清除率:
ABTS自由基清除率=(A0-A)/A0×100%
(11)
1.3.6.4 油脂氧化抑制作用
参照 Liu等[9]方法略作修改。先制备亚油酸体系:500 μL亚油酸溶于30 mL 0.2 mol/L pH为7.0的PBS并超声波10 min;再称取1.5 g TCA,0.038 g TBA,0.21 mL浓盐酸分别溶于10 mL蒸馏水中,用来制备TCA-TBA-HCl溶液。
在试管分别依次加入2 mL的样品溶液、2 mL亚油酸分散体系和2 mL 0.4 mmol/L的FeSO4溶液, 在37 ℃水浴锅中避光反应1 h,再向试管中加入4 mL 配好的TCA-TBA-HCl溶液,混匀后煮沸15 min, 冷却后4 500 r/min离心10 min,在532 nm处测上清液的吸光度为As。对照组用蒸馏水取代PPIHs溶液,吸光度为Ac。按照式(12)计算油脂氧化抑制率:
油脂氧化抑制率=(AC-AS)/AC×100%
(12)
1.3.7 不同加工条件对PPIHs抗氧化能力稳定性的影响
1.3.7.1 pH对抗氧化能力的影响
参照Gao等[12]方法略作修改,将PPIHs分别溶于pH为2、4、6、8、10的PBS缓冲溶液中配制成质量浓度为5 mg/mL的样品溶液,放于4 ℃静置1 h后,测定其体外抗氧化能力的变化趋势。
1.3.7.2 温度对抗氧化能力的影响
参照Yu等[13]方法略作修改,将质量浓度为5 mg/mL的PPIHs溶液,在20、40、60、80、100 ℃下保温1 h后,测定其体外抗氧化能力的变化趋势。
1.3.8 平均粒径和Zeta电位的测定
将样品配制成1 mg/mL的溶液以待测定。在石英比色皿样品池中加入一定的待测液,并使其无气泡。放入Zeta电位与粒度分析仪,设置相应参数,等待120 s至样品温度达到平衡(25 ℃),开始测定样品的粒径分布和Zeta电位。
1.3.9 紫外吸收光谱测定
参照郑召君等[14]方法并略作修改。将样品溶于蒸馏水中配制成浓度为0.10 mg/mL的溶液,以蒸馏水作为空白对照,扫描波长范围为200~380 nm,记录紫外扫描图谱。
1.3.10 傅里叶红外光谱测定
参照曾琪等[15]方法并略作修改。称取10 mg样品与1 g的溴化钾粉末混合后压片,把红外光谱仪分辨率设置为 4 cm-1,扫描次数设置为32次,扫描波数谱段范围设置为400~4 000 cm-1,再进行测定。
1.3.11 外源性荧光光谱测定
参考Evangelho等[16]方法并略作修改。先将样品溶于10 mmol/L磷酸盐缓冲液(pH 7.0)配制成质量浓度为1 μg/mL的溶液,而后在1.5 mL样品稀释溶液中加入20 μL 8 mmol/L 8-苯氨基-1-萘磺酸(ANS),将荧光分光光度计的扫描波长范围设置成400~700 nm,激发波长为390 nm,狭缝宽度设置成5.0 nm后进行扫描。
1.3.12 数据分析
所有实验均重复3次,数据均以平均数±标准差表示,实验分析采用Origin 8.5软件对数据进行图表处理;统计分析应用 SPSS 19.0 软件,多组均数比较采用单因素方差分析中Ducan进行显著性分析,以P<0.05表示两者之前存在显著差异。
2 结果与分析
2.1 PPIHs的理化性质
PPIHs的溶解度、起泡性和泡沫稳定性以及乳化性和乳化稳定性如表1所示。PPIHs在pH为2~10范围内具有良好的溶解性,与前期研究中获得PPIs相比[9],溶解度增加了10%~20%。PPIHs的起泡性较好,但在pH=6时起泡性急剧降低,可能因为此时蛋白质之间的静电斥力增加,阻碍了蛋白质在空气-水界面的吸附[17]。无论在酸性环境下还是碱性环境中,酶解产物的泡沫稳定性很低,可能是因为在酸性或碱性条件下,具有相同电荷类型的离子浓度增加,由于肽通过离子之间的排斥作用,以及微观肽无法保持稳定的泡沫使蛋白质水解物的泡沫稳定性下降[18]。
通过乳化性和乳化稳定性可以表征蛋白质的乳化性能,蛋白质在水/油混合过程中吸附在油滴界面降低其界面张力,乳化作用的大小主要由蛋白质分子链内部分布的亲水或疏水基团所决定的[19]。PPIHs在不同pH条件下的乳化性和乳化稳定性存在显著差异(P<0.05)。与PPIs[9]相比,酶解后蛋白质的乳化性和乳化稳定性得到明显的改善。可能因为在酶解后,蛋白质结构发生改变,具有较高溶解度和较低分子尺寸的水解产物能在油-水界面中扩散和扩散,此外,暴露的疏水基团改善了脂质和蛋白质之间的相互作用,使水解产物的乳化性增强[20]。
2.2 PPIHs的抗氧化稳定性
2.2.1 温度对PPIHs抗氧化能力的影响
食品加工过程中,热处理是最常见的方法之一,讨论PPIHs 的耐热性对其应用具有重要的意义。图1分析了PPIHs 经过不同温度(20~100 ℃)保温1 h后其抗氧化活性的变化。结果表明,温度在20~80 ℃区间时,PPIHs的抗氧化能力稳定性比较好;当温度高于80 ℃时,其抗氧化稳定性显著降低。加热处理在一定程度上改变了抗氧化肽的空间结构,从而导致样品的抗氧化能力发生变化[21]。
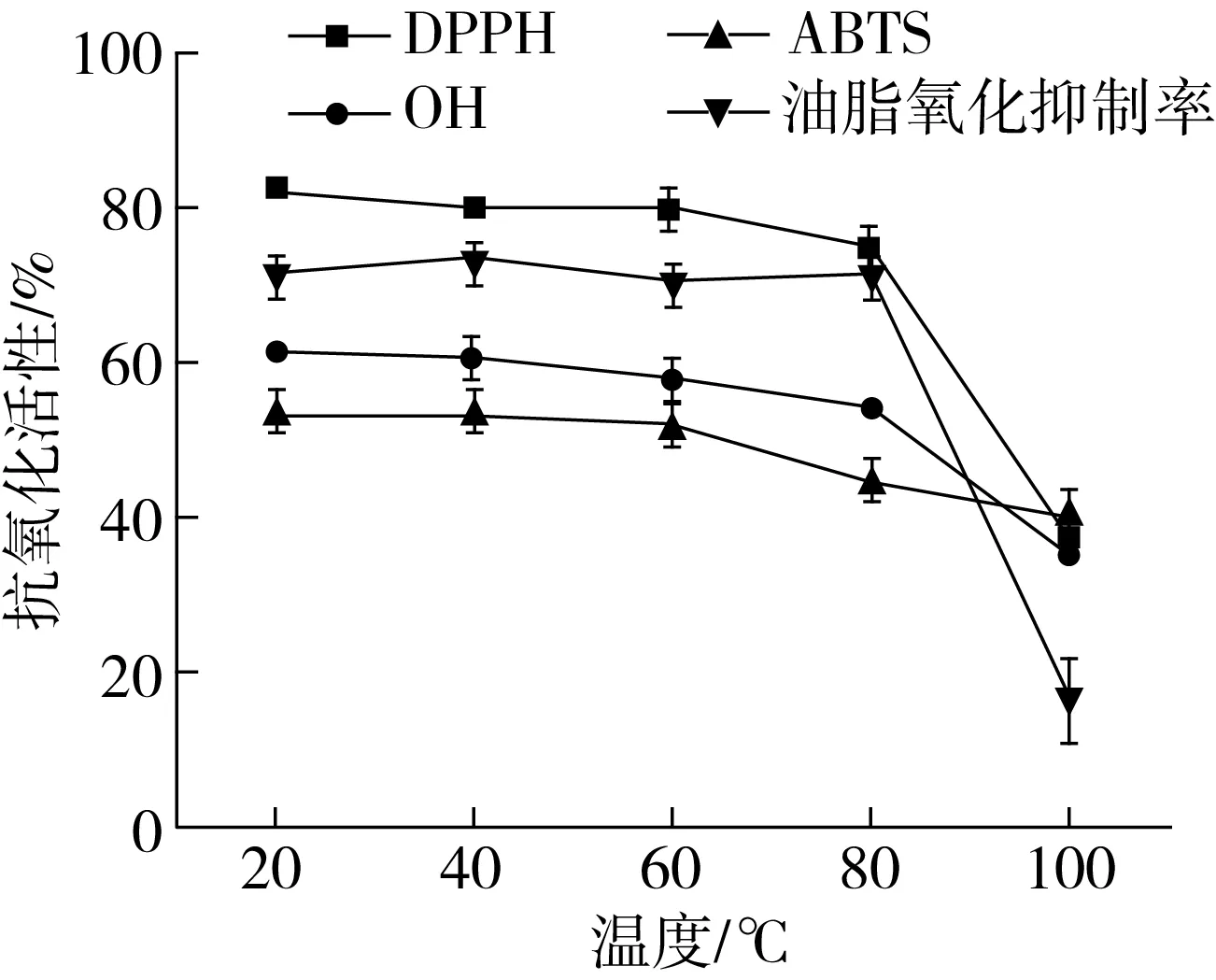
图1 温度对PPIHs抗氧化活性的影响
2.2.2 pH对PPIHs抗氧化能力的影响
pH对PPIHs抗氧化稳定性的影响如图2所示。PPIHs对DPPH的清除能力在pH为2~8范围内稳定,大于8以后急剧下降。可能是因为在碱性条件下肽发生消旋或者脱酰胺反应导致肽链构象发生变化,从而影响其抗氧化活性;而在酸性环境中,肽的极性氨基酸会暴露出来,阻碍疏水性氨基酸的反应[22]。PPIHs在脂质氧化体系中的抗氧化能力及对-OH自由基的清除能力在pH 2~10范围内比较稳定。而酶解产物在酸性条件下对ABTS自由基的清除能力比较差,但在碱性条件下比较好,这与玉米抗氧化肽[23]的结果相似,而牛慧慧等[24]对pH为蛋清抗氧化肽的影响进行研究,发现在碱性条件下不利于蛋清抗氧化肽的抗氧化活性的保持。

图2 pH对PPIHs抗氧化活性的影响
2.3 酶解产物的超滤分离
由图3a可以看出经酶解后PPIHs条带增多,分子质量组分比较复杂,进行纯化比较困难。为了研究分子量大小与结构之间的区别,以及对其抗氧化功能的影响,通过超滤技术将酶解产物分成PPIHs-Ⅰ、PPIHs-Ⅱ和PPIHs-Ⅲ3种不同分子质量大小的组分。与PPIs相比,PPIHs的分子质量变小,说明分子质量大的蛋白被水解,同时产生分子质量在6.5~14.4 ku的酶解产物,可能下面还存在一些小于3.3 ku的小分子肽段没有跑出来条带。超滤后,大于10 ku酶解产物的分子质量在10.0 ku以上;3~10 ku组分的电泳图中的条带分别在14.4 ku以下;<3 ku的超滤组分在Tricine-SDS-PAGE电泳图中体现不出来。图3b为超滤后各组分的提取率,其中PPIHs-Ⅰ的提取率最高为42%,其次是PPIHs-Ⅲ、PPIHs-Ⅱ。
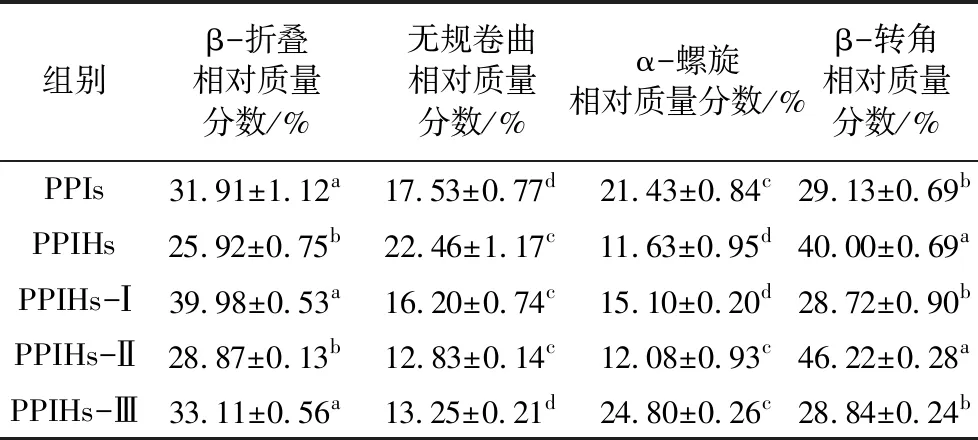
表2 酰胺Ⅰ带拟合PPIs、PPIHs、PPIHs-Ⅰ、PPIHs-Ⅱ和PPIHs-Ⅲ的二级结构组成
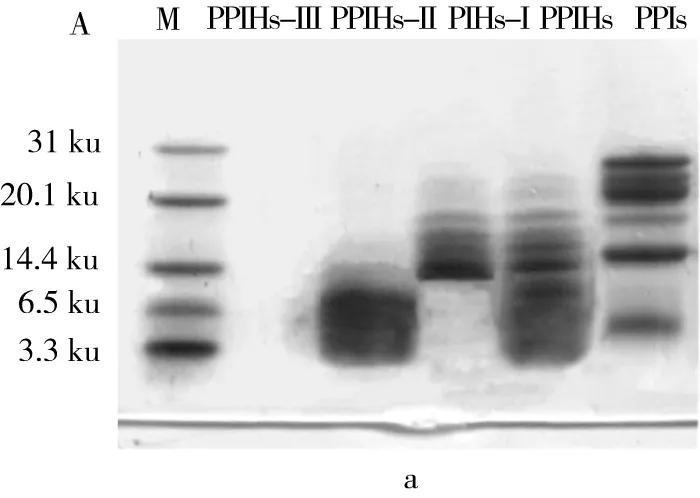
注:图3中不同小写字母表示差异显著(P<0.05),余同。
2.4 超滤各组分的粒径分布和平均粒径
图4为超滤各组分的粒径分布曲线和平均粒径图。结果表明,PPIs的平均粒径为(1 071.7±18.3)nm,在粒径分布中可以观察到2个峰,一个较小的峰分布在100~1 000 nm之间,另一个较大的峰分布在1 000~10 000 nm之间。相比之下,酶解将蛋白质转化为分子尺度较小的蛋白质、肽或氨基酸,PPIHs的平均粒径为(423.3±3.9)nm,显著(P<0.05)降低至纳米级,其粒径分布在100~10 000 nm之间,其主峰分布在600~700 nm,有一个小峰分布4 000~6 000 nm之间,可能是未水解的蛋白;与PPIHs 相比,PPIHs-Ⅱ 和PPIHs-Ⅲ的平均粒径显著(P<0.05)下降,而PPIHs-Ⅰ的平均粒径显著增加,可能因为分子量较大的蛋白集中出现在此部分,导致平均粒径略有上升。经过酶解和超滤,PPIs的平均粒径显著降低,可能会促进肠道的吸收,使其功能性得到充分发挥,满足人体对其持续需求[25]。
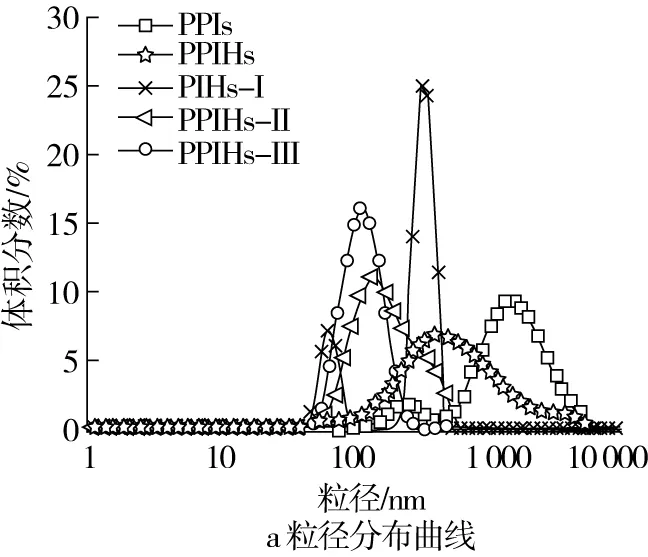
图4 超滤各组分的粒径分布曲线和平均粒径
2.5 超滤各组分的Zeta电位
蛋白质的Zeta电位是对水溶液中蛋白质的表面净电荷的表征,这种表面净电荷是由于蛋白质表面官能团的电离或者从溶液中吸附到蛋白质表面的离子引起的[26]。图5为超滤后各组分的Zeta电位变化情况。超滤后各组分的Zeta电位均为负值,说明其表面均带有负电荷。这可能与蛋白表面暴露出可电离氨基和羧基的数量变化有关,如燕麦蛋白、牡丹籽蛋白和鹰嘴豆分离蛋白的水解物[14,27,28]中也有相似的结果。与PPIs相比,PPIHs的Zeta电位绝对值略有提高,水解过程中释放的带负电荷的氨基酸略多于带正电荷的氨基酸,可能是导致起泡性较好的原因。与PPIHs相比,PPIHs-I的 Zeta电位的绝对值显著增加,PPIHs-Ⅲ的 Zeta电位的绝对值无显著变化,而PPIHs-Ⅱ电位的绝对值显著下降(P<0.05)。Zeta电位的绝对值越大表明有大量的静电斥力使颗粒之间不易发生聚集,颗粒呈散落分布状态[29],会提高PPIHs-Ⅰ溶液的稳定性。
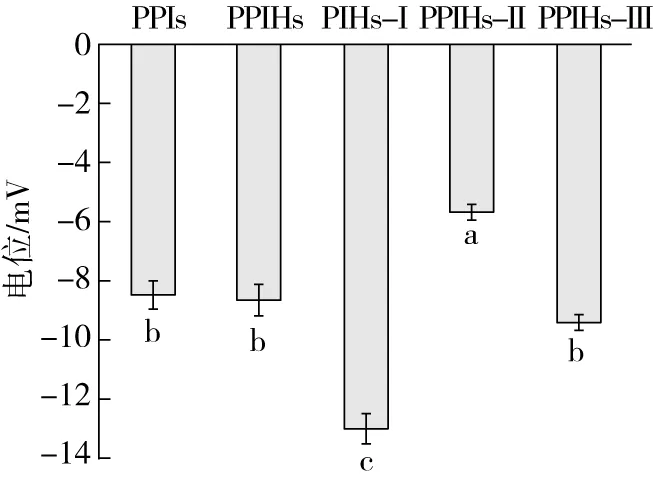
图5 各组分的Zeta电位
2.6 超滤各组分的抗氧化活性
超滤后各组分的抗氧化活性如图6所示。与PPIs相比,PPIHs的4种抗氧化指标均显著提高(P<0.05),可能由于酶解打开并破坏蛋白质的天然结构,暴露出更多能与氧化剂反应的活性肽或者氨基酸[5]。在PPIHs各组分中,PPIHs-Ⅰ对ABTS自由基的清除能力最强,可能由于具有清除ABTS自由基能力的物质大部分存在PPIHs-Ⅰ中;PPIHs-Ⅱ的油脂氧化抑制能力以以及-OH和DPPH自由基的清除能力较强,这可能是因为PPIHs-Ⅱ中暴露出较多的疏水性氨基酸残基,可以接触并捕捉疏水性自由基[30]。
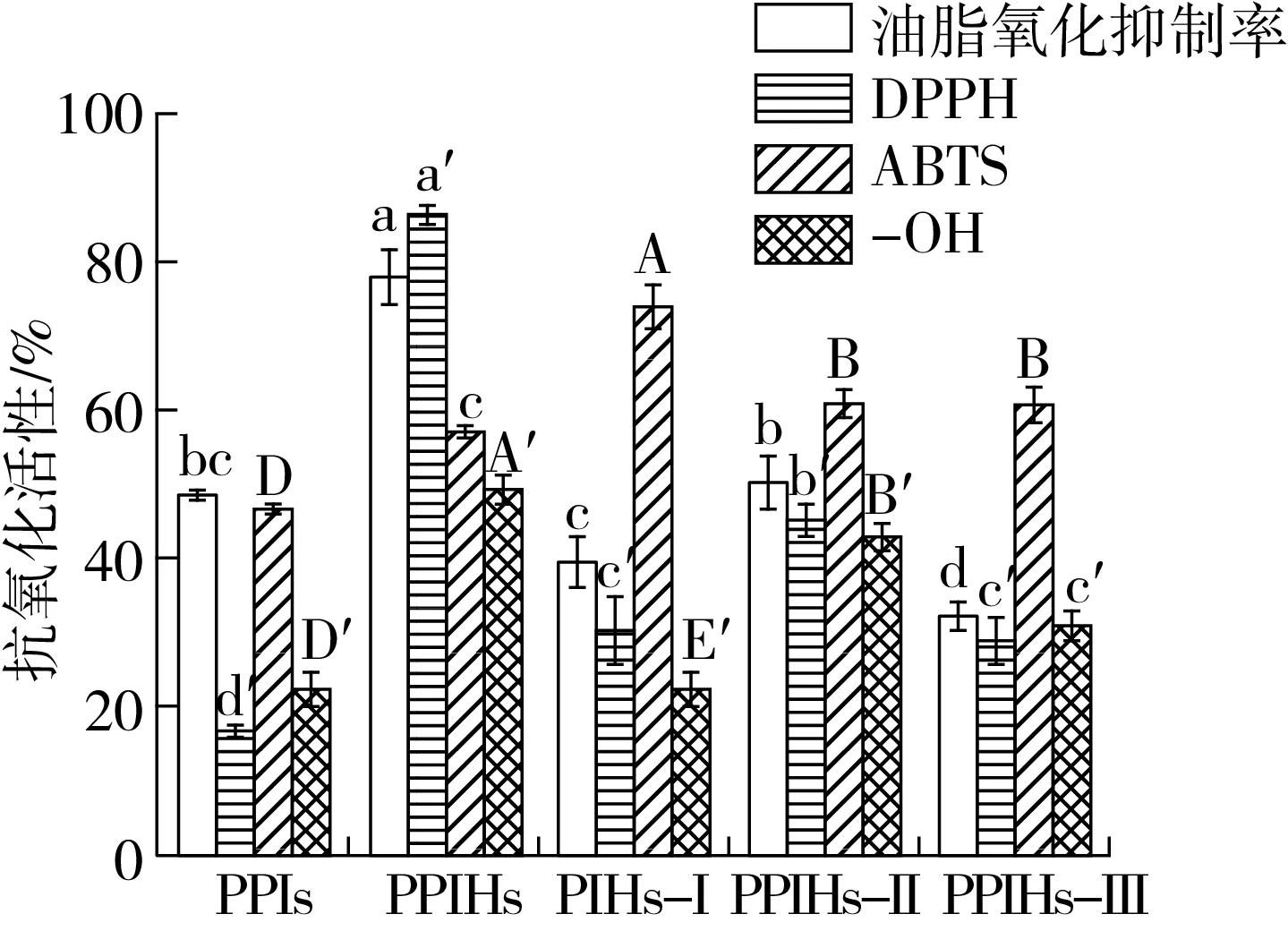
图6 各组分的Zeta电位
2.7 超滤各组分的紫外全波长扫描

图7 PPIs、PPIHs、PPIHs-Ⅰ、PPIHs-Ⅱ和PPIHs-Ⅲ的紫外波长扫描
2.8 超滤各组分的傅里叶红外光谱
傅里叶变换红外光谱(FTIR)可反映蛋白质的官能团和二级结构的变化,蛋白质的二级结构在1 600~1 700 cm-1之间,与酰胺Ⅰ带重叠,使用peakfit软件拟合出在酰胺-Ⅰ区域的峰位,根据峰的中心位置将其分配到相应的结构中,1 600~1 639 cm-1谱带属于β-折叠;1 640~1 650 cm-1谱带属于无规卷曲;1 651~1 660 cm-1谱带属于α-螺旋;1 661~1 700 cm-1谱带属于β-转角[33]。
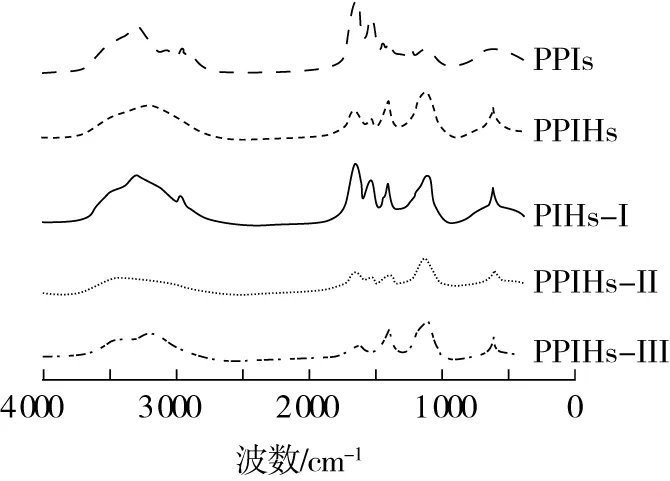
图8 PPIs、PPIHs、PPIHs-Ⅰ、PPIHs-Ⅱ和PPIHs-Ⅲ的傅里叶红外光谱
选取1 600~1 700 cm-1波段进行曲线拟合,经去卷积化处理,得到PPIs、PPIHs以及PPIHs-Ⅰ、PPIHs-Ⅱ和PPIHs-Ⅲ的酰胺-Ⅰ带图谱,对其进行二级结构成分分析,得到蛋白二级结构的定量信息,见表2。PPIs的二级结构主要以β-折叠和β-转角为主,相对质量分数分别为31.91%和29.13%。经酶解后,PPIHs的二级结构以无规卷曲和β-转角为主。与PPIs相比,PPIHs的无规卷曲和β-转角相对含量提高,β-折叠含量和α-螺旋相对减少。蛋白经酶解后由有序变得无序,稳定蛋白二级结构的氢键和静电作用发生变化,使其空间结构遭到破坏[34],这也可能是抗氧化活性提高的原因。与PPIHs相比,PPIHs-Ⅰ、PPIHs-Ⅱ和PPIHs-Ⅲ的β-折叠和α-螺旋增加,无规卷曲减少;PPIHs-Ⅱ的β-转角增加,而PPIHs-Ⅰ和PPIHs-Ⅲ的β-转角减少。
2.9 超滤各组分的荧光光谱
蛋白质的疏水性残基大部分位于蛋白质内部,酶解使蛋白分子展开,暴露出更多疏水性氨基酸,导致蛋白质的表面疏水性发生变化[35]。由图9可见,与 PPIs 相比,PPIHs 荧光强度增加,这表明经过酶解处理后蛋白质分子内部振动增强,维系蛋白空间结构的次级键遭到破坏,暴露出蛋白质内部的疏水性氨基酸残基,导致疏水性增强。正是由于更多的疏水性氨基酸残基存在,才能使其在均质的过程中迅速扩散并瞬间吸附到新形成的油滴表面,进而增加水解产物的乳化性[30]。与PPIHs相比,PPIHs-Ⅰ 的荧光强度下降,可能因为大部分疏水性氨基酸残基存在于分子量较小的酶解物中,而 PPIHs-Ⅱ 和 PPIHs-Ⅲ 的荧光强度增加,进一步说明小于10 ku酶解物中含有更多的疏水性氨基酸残基。
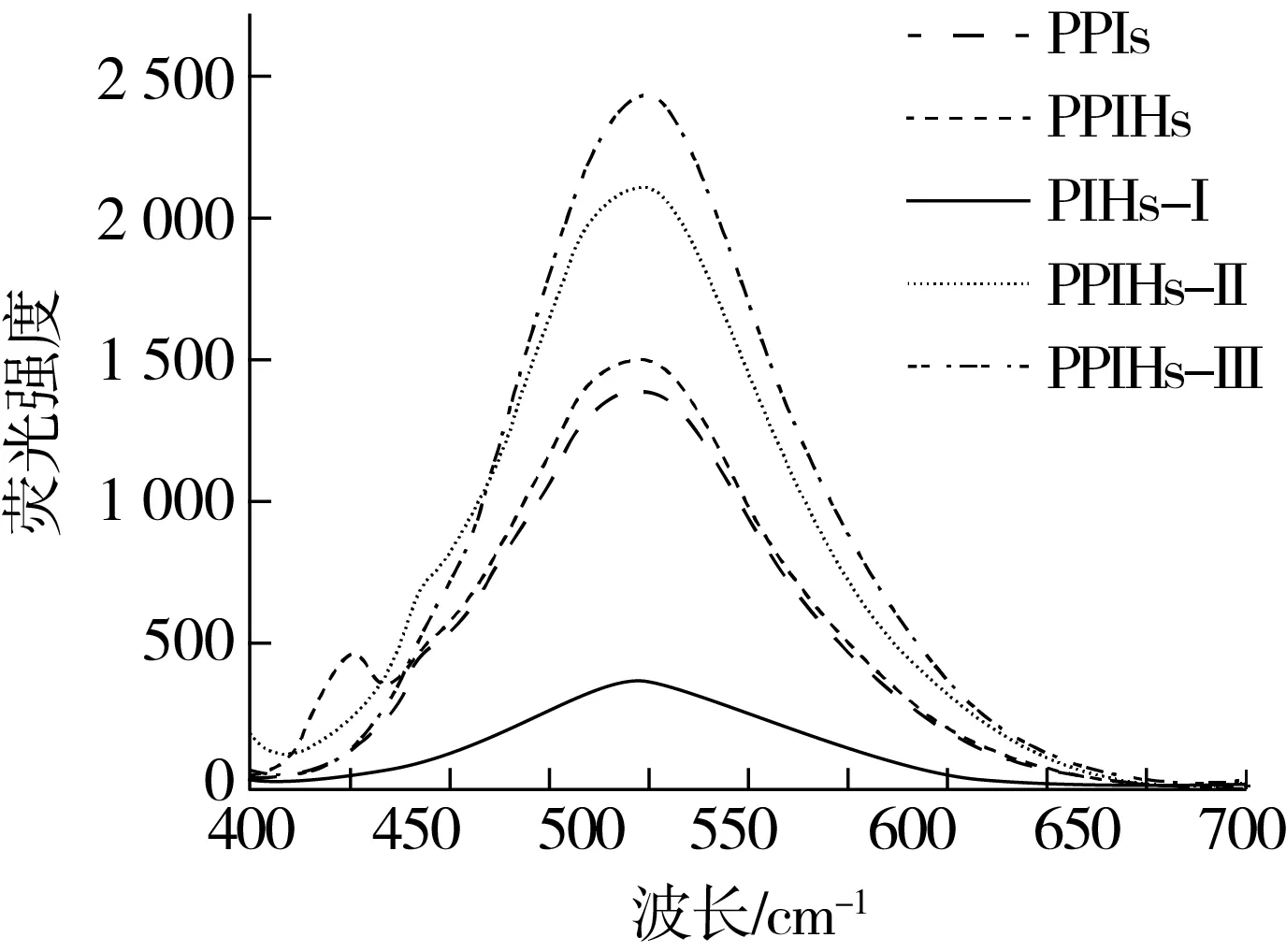
图9 PPIs、PPIHs、PPIHs-Ⅰ、PPIHs-Ⅱ和PPIHs-Ⅲ外源性荧光光谱
3 结论
利用超声辅助蛋白酶K对马铃薯蛋白酶抑制剂进行酶解,系统表征了水解产物的理化性质以及pH和温度对其抗氧化稳定性的影响,并讨论了超滤后各组分的抗氧化能力和结构特性。酶解后,PPIHs的分子质量由大分子蛋白向小分子肽转变,溶解度增加,抗氧化稳定性增强,酶解后紫外吸收光谱发生红移,蛋白质的构象改变,疏水性增加,β-折叠和α-螺旋降低,无规卷曲和β-转角增加。超滤后PPIHs-Ⅰ的平均粒径和Zeta电位的绝对值略有增加,对ABTS自由基的清除能力最强,荧光光谱显著下降;PPIHs-Ⅱ 的油脂氧化抑制能力以及对DPPH和OH自由基的清除能力较强,PPIHs-Ⅲ 的抗氧化能力较弱,其荧光光谱增强,二级结构发生改变。
猜你喜欢
煤气与热力(2021年12期)2022-01-19
成都大学学报(自然科学版)(2021年1期)2021-05-22
当代水产(2020年4期)2020-06-16
当代水产(2020年3期)2020-06-15
石油沥青(2019年3期)2019-07-16
中成药(2018年8期)2018-08-29
科学中国人(2018年8期)2018-07-23
石油沥青(2018年3期)2018-07-14
中成药(2018年2期)2018-05-09
中国公路(2017年17期)2017-11-09
