
基于单细胞测序探索METTL1与胃癌恶性进展的关联性研究
2023-11-04 02:49喻大军李靖王彬彬李志祥王凯杨杰
右江民族医学院学报 2023年5期
喻大军,李靖,王彬彬,李志祥,王凯,杨杰
(蚌埠医学院第一附属医院肿瘤外科,安徽 蚌埠 233004)
胃癌是世界上第五常见的癌症,也是第三常见的癌症相关死亡原因[1]。而胃癌的发病率逐年增高,已经成了危害中国人民生命健康的威胁之一[2-3]。尽管系统治疗包括病理方法和分子靶向药物的生物标志物检测,但5年生存率仅为31.5%[4]。因此,寻找一种生物标志物来提高临床疗效显得十分必要。单细胞RNA测序,也被称为scRNA-seq,是一种将组织分解成单个细胞以区分肿瘤细胞和非癌细胞并分析表达模式以推断亚克隆的方法[5]。scRNA-seq是一种有潜力用于研究转移性癌细胞的方法,从原始位置转移的癌细胞的整体水平表达模式会因其扩散到的局部组织而改变,而通过scRNA-seq探索癌症发病因素,已经成了研究热点[6-7],甲基转移酶-1蛋白(methyltransferase like-1 protein,METTL1)是一种甲基转移酶,可催化真核生物mRNA的n7 -甲基鸟苷(m7G)修饰[8]。越来越多的证据揭示了METTL1的致癌潜力[9]。然而METTL1在胃癌中的作用少见报道,因此结合单细胞测序结果并探索METTL1的功能显得十分关键。
1 材料和方法
1.1 scRNA-seq数据采集及预处理 从Gene Expression Omnibus(GEO,http://www.ncbi.nlm.nih.gov/geo/)数据库中找到GSE163558数据集,收集6例胃癌患者10份样本共42 968个细胞的scRNA-seq数据,基于Illumina NovaSeq 6000,读取深度为10倍基因组学。首先,从样本中去除不符合以下质量控制要求的低质量细胞:①排除在少于3个细胞中发现的基因;②总检测基因少于200个的细胞被排除;③表达少于10%的线粒体基因的细胞被排除在外。使用线性回归模型来标准化遗留细胞的基因表达水平,采用主成分分析法进行降维。然后,使用均匀流形逼近和投影(UMAP)方法对所有单元进行聚类分类分析,并使用原始的15个pc对数据进行降维。在单细胞RNA-seq研究中,各组之间的差异表达基因(DEGs)是进行GSEA富集分析的基础。
1.2METTL1与随机森林模型构建 胃癌(n=433)的Bulk RNA测序(RNA-seq)从GEO数据库(GSE84437)中检索,通过IPA软件鉴定METTL1相关基因。采用了一种被称为随机森林(RF)方法的机器学习技术进行生存分析,以确定来自预后相关特征的哪些基因可能具有预后价值。RF方法涉及开发大量的决策树或分类树,然后将其用于对某个输入数据向量进行分类的任务。射频技术采用从初始数据集派生的各种自举和随机分裂样本,以构建每个单独的决策树。为了从预后相关特征中确定哪些基因可能具有预后意义,使用RF方法进行了生存分析。
1.3 RT-PCR实验 用Trizol(上海碧云天生物技术有限公司)从细胞中分离总RNA,并稀释到DNase/ RNase-free水中。定量后,使用RevertAid First Strand cDNA Synthesis Kit将总RNA(每个样品2 μg)逆转录为cDNA。最后使用SYBRGreen PCR Master Mix进行实时PCR检测目的基因的表达水平。以GAPDH作为对照。本研究引物序列购自生工生物工程(上海)股份有限公司,见表1。

表1 PCR序列信息
1.4 细胞培养和转染 从武汉细胞库获得GSE-1、AGS、MNK-45、HGC27和MGC-803细胞系,所有细胞在1640培养液[abs9484,爱必信(上海)生物科技有限公司)]中培养。构建沉默METTL1的小干扰RNA后,利用Lipofectamine 3000[L3000008,赛默飞世尔科技有限公司]转染载体。载体序列如下:正义:5′-CACCGCGGTAGTAGCGCTTCTGGGG-3′;反义:5′-AAACCCCCAGAAGCGCTACTACCGC-3′。
1.5 CCK-8实验 CCK-8试剂检测细胞增殖活性;将细胞接种于96孔板,密度为5 000个细胞/孔。在37 ℃和5%CO2的培养箱中培养。孵育1 d、2 d、3 d后,每孔加入10 μL CCK-8试剂。37 ℃孵育1~2 h后,在酶标仪上读取450 nm处的吸光度。
1.6 EDU实验 将各组处理后的细胞种植于12孔板中,随后按照EDU试剂盒(C0078S,上海碧云天生物技术有限公司)说明书,以RPIM1640培养基稀释成2X EDU工作液,37.5 ℃孵育。接下来,细胞洗涤3次,用4%多聚甲醛处理并固定,用通透液(P0095,上海碧云天生物技术有限公司)重新洗涤3次。根据说明配制反应溶液,然后加入细胞中。用DAPI染色液配置细胞,在荧光显微镜下拍摄图像。
1.7 免疫组化 收集蚌埠医学院第一附属医院病理确诊为胃癌患者的癌旁和癌组织样本,并在病理科制作成免疫组化蜡块,随后把组织切成样片,取出切片后,取抗原,加入3%过氧化氢浸泡。在此切片上,快速加入免疫阻断溶液进行阻断,然后用一抗(14994-1-AP,武汉三鹰生物技术有限公司)进行处理。最后将盖片风干,在荧光显微镜下拍摄切片。
1.8 统计学方法 使用R(4.1.1)编程语言进行统计研究。采用Kaplan-Meier法和log-rank检验对不同组的总生存率进行分析和比较。单因素t检验用于评估两组间的差异。多组间差异分析采用单因素方差分析(ANOVA)。P<0.05认为差异有统计学意义。
2 结果
2.1 单细胞测序寻找癌细胞分区趋势 为了确定数据源的可靠性,首先分析了来自数据库(https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi)的胃癌单细胞数据。结果发现胃癌(gastric cancer,GC)单细胞序列数据可分为18组(见图1A)。10组和11组的肿瘤相关基因(EPCAM、CD24、CDH1、ELF3、KRT18、KRT19、KRT8和MUC1)的高表达,表明10组和11组的肿瘤细胞差异较大(见图1B)。分布图如图1C和热图如图1D所示。
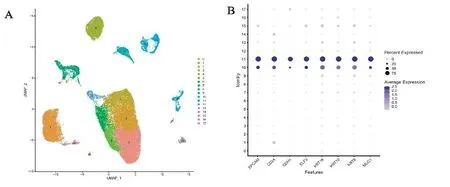
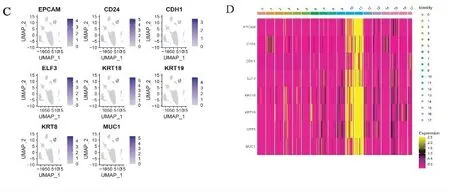
注:A.单细胞序列将胃癌组织划分为18个亚组;B.10/11亚群中表达的不同基因;C.这些差异基因的位置;D.差异基因表达的热图。
2.2 差异基因筛选 为了进一步确定肿瘤中异常表达的基因,本研究使用基因集富集分析(gene set enrichment analysis,GSEA)来比较肿瘤和正常组织之间的差异。研究结果发现,与正常组织相比,肿瘤组织中tRNA相关信号通路活跃(见图2A)。同时,也从GSE84437的肿瘤组织中发现了METTL1基因过表达(见图2B和2C)。生存分析显示,METTL1高表达的患者比METTL1低表达的患者生存率更高(见图2D)。
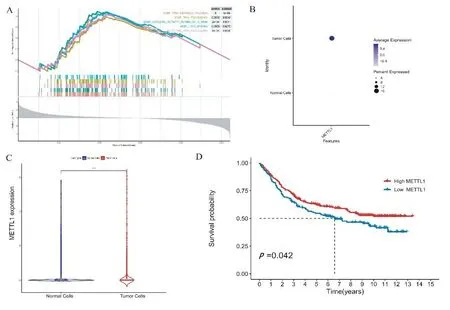
注:A.基因集富集分析显示tRNA相关通路存在差异表达;B.METTL1在GC细胞中的表达较高;C.METTL1在肿瘤细胞和正常细胞间的表达情况;D.METTL1在不同表达水平下的生存曲线。
2.3 差异基因构建RF模型并检测效率 接下来,为了进一步发现潜在的调控机制,本研究通过独创性途径分析(ingenuity pathway analysis,IPA)发现METTL1相关基因,挑选出预后基因并在网络图中呈现(见图3A、图3B)。为了更好地研究模型,根据训练组中最佳截断值将患者分为高危组和低危组(见图4A)。同样的方法也适用于试验组(见图4B)和所有数据集患者(见图4C)。与死亡风险较低的患者相比,死亡风险较高的个体预后较差(见图4D~图4F)。在高风险患者和低风险患者之间发现的预后相关基因的表达变化以热图的形式呈现(见图4G~图4I)。
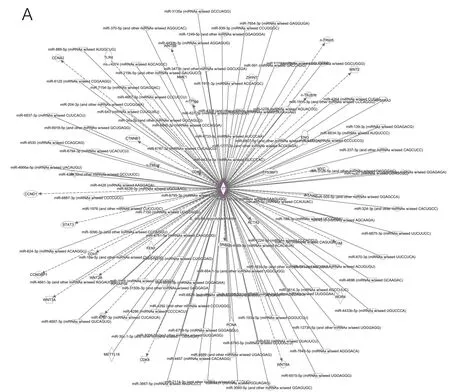
注:A.METTL1及其相关基因的网络;B.相关预后基因网络。

注:A.训练组、B.试验组和C全患者的风险评分分布及中位数;D.训练组、E.试验组和F全患者的OS状态、OS和风险评分分布;G.训练组、H.试验组和I全组患者高危、低危预后基因热图。
2.4 沉默METTL1抑制了胃癌细胞增殖 免疫组化(immunohistochemistry,IHC)结果显示,METTL1在胃癌组织中表达升高(见图5A)。RT-PCR检测显示METTL1在AGS中表达最高(见图5B),随后PCR检测沉默载体构建效率(见图5C)。随后CCK-8、克隆实验和EDU实验结果显示,相较于NC组,si-METTL1组的增殖效率降低,且差异具有统计学意义(P<0.05),见图5D~图5F。
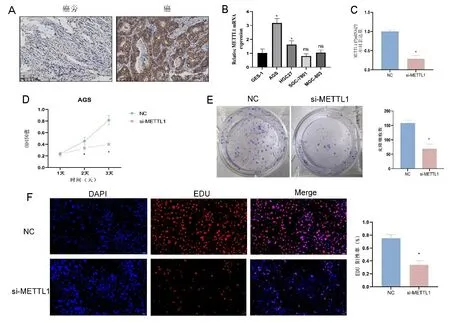
注:A.免疫组化检测METTL1在胃癌患者癌与癌旁组织的表达;B.RT-PCR检测METTL1在胃癌细胞系中的表达;C.RT-PCR检测沉默METTL1载体的构建效率;D~F.CCK-8、细胞克隆和EDU实验检测NC组和si-METTL1组中AGS细胞的增殖情况。*P<0.05。
3 讨论
研究表明,癌症的异质性是癌症诊断和治疗的重要问题[10],scRNA-seq领域的最新发展使得检测源自原代组织的异常细胞间相互作用、化疗耐药性和免疫抑制微环境成为可能,这些都是癌症研究的重要方面。原发性乳腺癌细胞从肿瘤细胞和免疫细胞中分离出来,并对实验结果进行比较,并构建有效的预后模型[11]。研究人员发现原发性胶质母细胞瘤存在较大的特异的异质性分化,表明存在不同的调控信号和治疗方案的可能[12]。KIM B S等[13]收集的scRNA-seq数据将肿瘤内SNV KRASG12D与肺腺癌细胞的表达异质性联系起来,这就能够在抗癌治疗反应的背景下解释亚群。就肿瘤内部的癌细胞而言,聚类分析结果显示了大量的GC异质性。
在这项研究中使用单细胞测序来筛选GC患者的差异表达基因。同时,基于METTL1相关预后基因构建随机森林预后模型,并通过随机森林模型预测预后基因与肿瘤预后关系,作为构建预后风险评分模型的基础。在训练集和测试集进一步验证了该模型,发现在预测患者具有较好的风险评分,从而证实了该模型良好的预测能力。METTL1可作为预测肿瘤预后反应的一种新的生物标志物。
METTL1是一种tRNA和miRNA修饰酶,在哺乳动物细胞中催化tRNA和miRNA的n7-甲基鸟苷(m7G)修饰[14-15]。近年来,有多项研究阐明了METTL1在人类恶性肿瘤中的作用。既往研究表明,METTL1在肝癌、结直肠癌、肺癌等人类恶性肿瘤中发挥着重要的生物学功能,例如METTL1在肝细胞癌中显示出致癌活性,而在结直肠癌中,它作为肿瘤抑制因子。此外,METTL1的过表达也通过调节miR-149-3p/S100A4/p53轴增加结直肠癌细胞对顺铂的化疗敏感性[16-18]。这些结果表明[19],维持高水平的功能性tRNA可能对METTL1在癌细胞中的作用至关重要。本研究中发现METTL1在胃癌AGS和HGC27细胞中高表达,且抑制METTL1后抑制了胃癌细胞增殖过程。既往研究表明[19],通过全基因组CRISPR-Cas9筛选,METTL1在胃癌中也有显著表达,具有成为治疗该疾病的治疗靶点的潜力。因此,METTL1作为一种新的标志物,在预测GC患者预后情况有重要价值,但具体的作用机制有待进一步研究。
猜你喜欢
新民周刊(2022年27期)2022-08-01
传染病信息(2021年6期)2021-02-12
科学(2020年4期)2020-11-26
中国卫生标准管理(2015年3期)2016-01-14
医学研究杂志(2015年9期)2015-07-01
中国当代医药(2015年20期)2015-03-01
生殖医学杂志(2015年11期)2015-02-28
生物医学工程学进展(2015年1期)2015-02-28
化学工业与工程(2015年1期)2015-02-10
中国中医药现代远程教育(2014年22期)2014-03-01
