
加味归芪片质量标准研究
2023-10-28 13:20王娟弟郭晓霞李冬华郭朝晖
安徽农业科学 2023年19期
王娟弟,李 敏,刘 蕊,郭晓霞,李 欣,李冬华,马 潇*,郭朝晖
(1.甘肃省药品检验研究院,甘肃兰州 730070;2.国家药品监督管理局中药材及饮片质量控制重点实验室,甘肃兰州 730070)
加味归芪片收载于1992年出版的卫生部药品标准《中药成方制剂》第六册(WS3-B-1118-92),具有补气、养血之功效,用于气血两亏、气虚体弱、肢体劳倦之症[1],该制剂疗效明确、适用人群广、副作用小;其处方中的3味药材当归、黄芪、党参均属于甘肃省道地药材。当归是伞形科植物当归[Angelicasinensis(Oliv.)Diels]的干燥根,具有补血活血等功效,主要分布在甘肃、四川及云南等地,甘肃岷县被誉为“千年药乡”,其产当归品质优良,又被称为岷归;其主要含有挥发油、有机酸、多糖和黄酮等成分[2-8]。2020年版《中国药典》[9]以阿魏酸作为评价当归药材和饮片质量的指标性成分之一。黄芪是豆科植物蒙古黄芪[AstragalusmembranaceusBge.var.mongholicus(Bge.) Hsiao]和膜荚黄芪[A.membranaceus(Fisch.) Bge.]的根,具有补气固表、利尿脱毒、排脓、敛疮生肌的功效,主要分布在甘肃、内蒙古和山西等地;其主要含有多糖类、皂苷类、黄酮类、生物碱类等成分[10-15]。2020年版《中国药典》[9]以黄芪甲苷作为评价黄芪药材和饮片质量的指标性成分之一。
加味归芪片质量标准的内容简单,仅有性状、显微鉴别、片剂通则项,不能有效控制产品质量。为此,该研究拟对加味归芪片质量标准进行提升,提高产品质量,从而更加有效控制加味归芪片的质量,确保加味归芪片的疗效,为临床用药提供质量保证。
1 仪器与材料
1.1 仪器高效液相色谱仪(日立,型号Chromaste);电子天平(瑞士梅特勒公司,型号ME204、MS205DU);旋转蒸发仪(瑞士BUCHI公司,型号Rotavapor R-300);离心机(艾本德公司,型号5810R);薄层色谱点样仪(瑞士卡玛公司,型号ATS4)。
1.2 试药与样品当归(中国食品药品检定研究院,批号120927-201617);阿魏酸(中国食品药品检定研究院,批号110773-201614);黄芪甲苷(中国食品药品检定研究院,批号120974-21813);甲醇、乙腈均为色谱级;水为屈臣氏水;其余试剂均为分析纯。加味归芪片(批号180501、181101、190101、190701、191201、191202、191203、191204、200101、200102、200103、200104)来源于佛仁制药有限公司。
2 方法与结果
2.1 当归成分的薄层色谱鉴别取本品适量,去包衣,粉碎,精密称定粉末5 g,加乙醚20 mL,超声处理10 min,滤过,滤液蒸干,残渣加乙醇1 mL溶解,作为供试品溶液。另取当归阴性药材5 g、当归药材(原料)0.5 g、当归对照药材0.5 g,同法制成当归阴性对照溶液、当归药材(原料)和当归对照药材溶液。按照薄层色谱法(通则0502)试验,吸取上述3种溶液各10 μL,分别点于同一硅胶G薄层板上,以环己烷-乙酸乙酯(4∶1)为展开剂,展开,取出,晾干。置紫外光灯(365 nm)下检视,结果见图1。
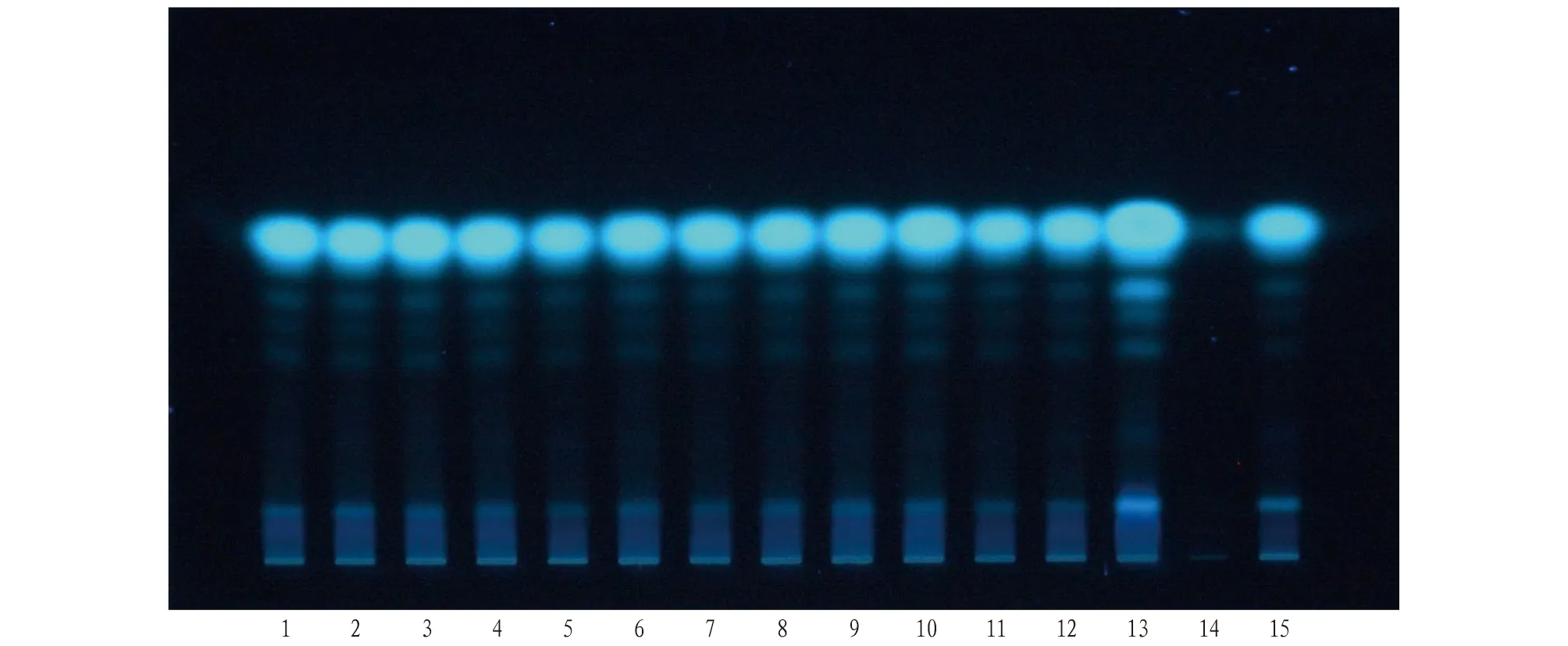
注:1~12为样品,批号依次为180501、181101、190101、190701、191201、191202、191203、191204、200101、200102、200103、200104;13为当归药材(原料);14为当归阴性样品;15为当归对照药材。Note:1-12 are samples, with batch numbers 180501, 181101, 190101, 190701, 191201, 191202, 191203, 191204, 200101, 200102, 200103, and 200104 in sequence; 13 is Angelica sinensis medicinal material (raw material); 14 is a negative sample of Angelica sinensis; 15 is the reference medicinal material of Angelica sinensis.图1 加味归芪片中当归对照药材薄层鉴别Fig.1 Thin layer identification of Angelica sinensis reference medicinal material in Jiawei Guiqi tablets
取本品适量,去包衣,粉碎,精密称定粉末5 g,加1%碳酸氢钠溶液50 mL,超声处理10 min,6 000 r/min离心5 min,取上清液用稀盐酸调节pH至2~3;用乙醚振摇提取2次,每次20 mL,合并乙醚液,挥干,残渣加甲醇1 mL溶解,摇匀,作为供试品溶液。另取当归阴性药材5 g、当归药材(原料)0.5 g,同法制成当归阴性和当归药材(原料)对照溶液。另取阿魏酸对照品,加甲醇制成1 mg/mL溶液,作为对照品溶液。按照薄层色谱法(通则0502)试验,吸取上述3种溶液各10 μL,分别点于同一硅胶G薄层板上,以环己烷-乙酸乙酯-二氯甲烷-甲酸(4∶1∶1∶0.1)为展开剂,展开,取出,晾干,置紫外光灯(365 nm)下检视,结果见图2。
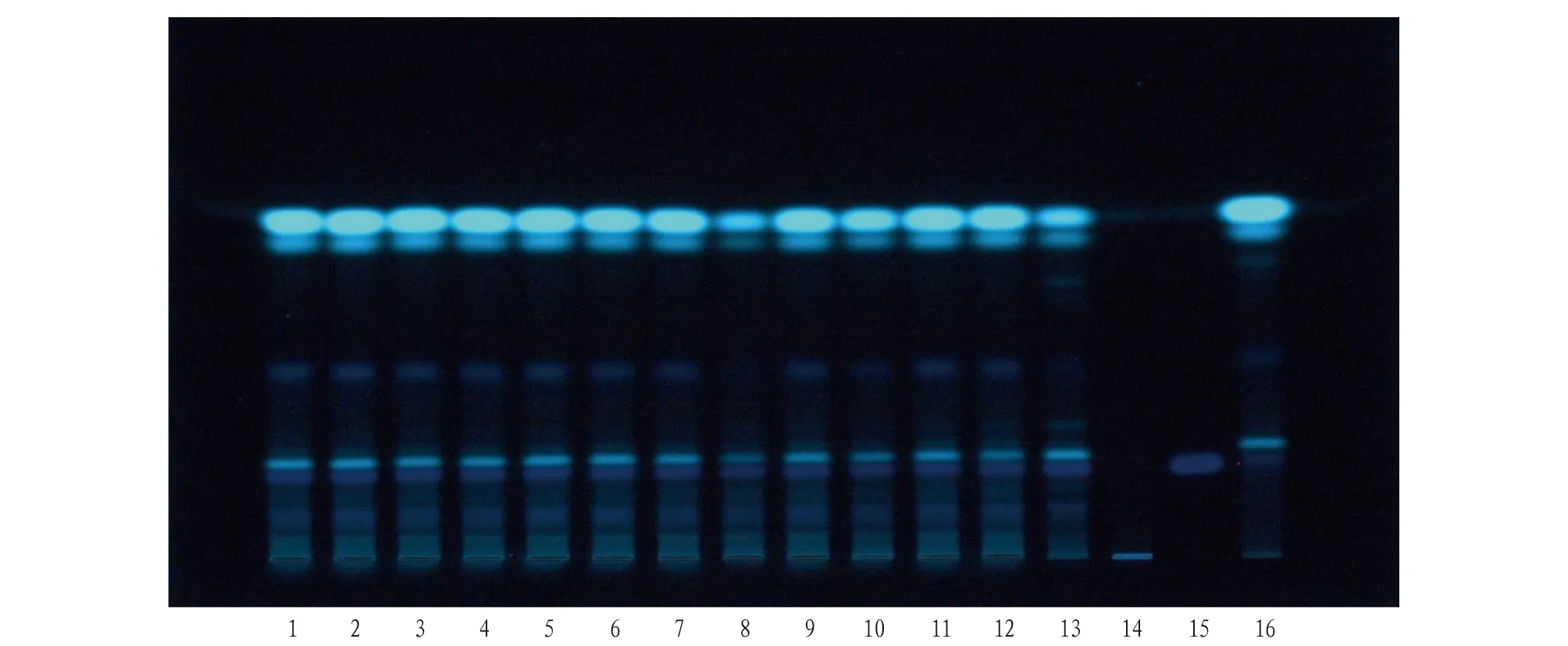
注:1~12为样品,批号依次为180501、181101、190101、190701、191201、191202、191203、191204、200101、200102、200103、200104;13为当归药材(原料);14为当归阴性样品;15为阿魏酸对照品;16为当归对照药材。Note:1-12 are samples, with batch numbers 180501, 181101, 190101, 190701, 191201, 191202, 191203, 191204, 200101, 200102, 200103, and 200104 in sequence; 13 is Angelica sinensis medicinal material (raw material); 14 is a negative sample of Angelica sinensis; 15 is the reference substance for ferulic acid; 16 is the reference medicinal material of Angelica sinensis.图2 加味归芪片中阿魏酸对照品薄层鉴别Fig.2 Thin layer identification of ferulic acid reference substance in Jiawei Guiqi tablets
加味归芪片样品、当归对照药材以及阿魏酸对照品均有相应的斑点,斑点清晰,分离条件好,并且加味归芪片当归阴性无干扰,因此对当归对照药材和阿魏酸对照品进行定性鉴别。
2.2 黄芪成分的薄层色谱鉴别取本品适量,去包衣,粉碎,精密称定粉末5 g,置锥形瓶中,加甲醇30 mL,超声处理30 min,放冷,过滤,收集滤液,蒸干,残渣溶解于10 mL 10%(V/V)氨水溶液中,待溶解完全,将溶液转移至分液漏斗中,用水饱和的正丁醇萃取3次(15、10、10 mL)。合并提取液,蒸干,残渣用甲醇溶解并转移至10 mL容量瓶中,加甲醇至刻度,即得。取黄芪对照药材1 g、黄芪药材(原料)1 g、黄芪阴性样品5 g,同法制备对照药材和黄芪药材(原料)溶液及阴性对照溶液。取黄芪甲苷对照品,加甲醇制成1 mg/mL溶液,即得对照品溶液。按照薄层色谱法(通则0502)试验,吸取上述溶液5 μL,分别点于同一硅胶G薄层板上,以三氯甲烷-乙酸乙酯-甲醇-水(2∶4∶2∶1)的下层溶液作为展开剂,取出,晾干,喷以10%硫酸乙醇,105 ℃干燥之显色,置紫外光(365 nm)下检视,结果见图3。
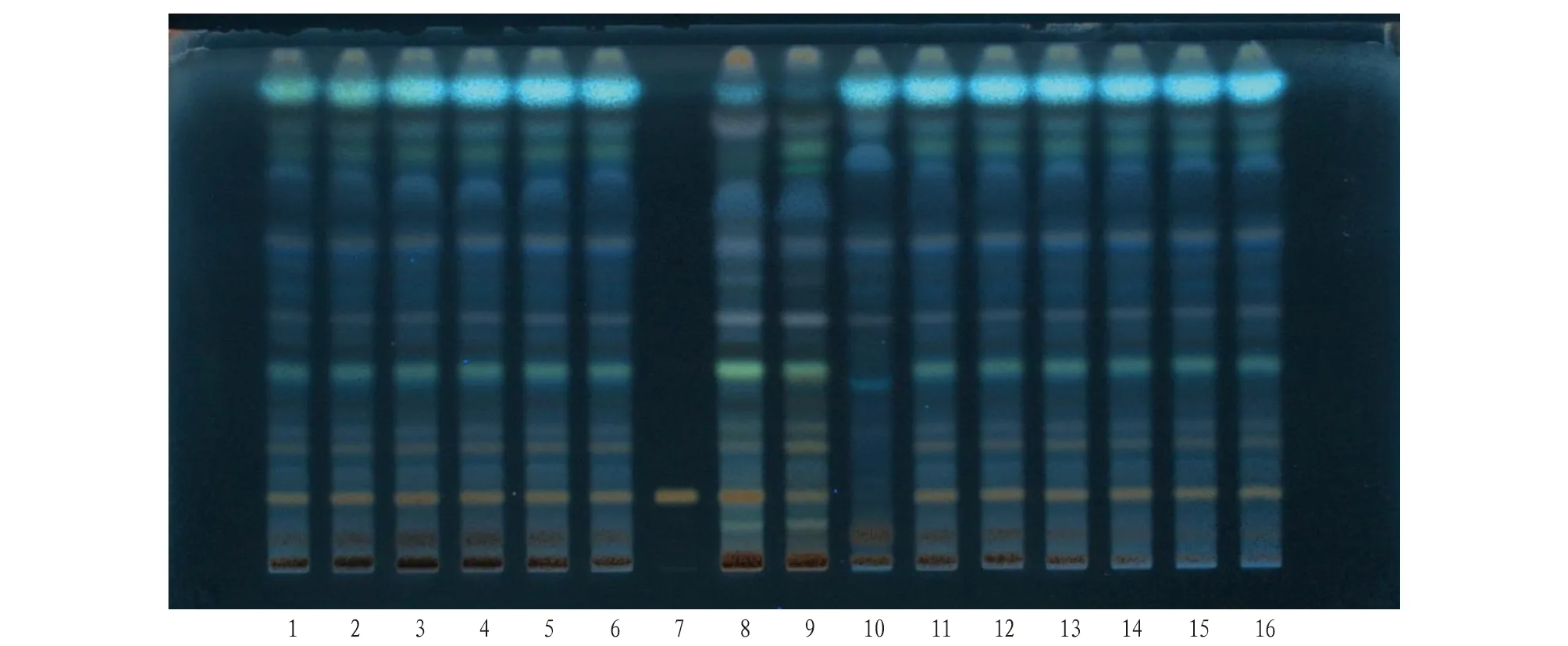
注:1~6为样品,批号依次为180501、181101、190101、190701、191201、191202;7为黄芪甲苷对照品;8为黄芪对照药材;9为黄芪药材(原料);10为阴性对照;11~16为样品,批号依次为191203、191204、200101、200102、200103、200104。Note:1-6 are samples, with batch numbers 180501, 181101, 190101, 190701, 191201, 191202 in sequence; 7 is the reference substance for astragaloside IV; 8 is the reference medicinal material of Astragalus membranaceus; 9 is Astragalus membranaceus medicinal material (raw material); 10 is a negative control; 11-16 are samples, with batch numbers 191203, 191204, 200101, 200102, 200103, and 200104 in sequence.图3 加味归芪片中黄芪甲苷对照品薄层鉴别Fig.3 Thin layer identification of astragaloside IV reference substance in Jiawei Guiqi tablets
加味归芪片样品与黄芪甲苷对照品有相应的斑点,斑点清晰,分离条件好,且黄芪阴性无干扰。加味归芪片样品与黄芪对照药材有相应的斑点,斑点清晰,但黄芪阴性有干扰。因此对黄芪甲苷对照品进行定性鉴别。
2.3 阿魏酸含量测定
2.3.1色谱条件。色谱柱为Agilent Eclipse Plus C18(4.6 mm×250 mm,5 μm);以乙腈-0.085%磷酸溶液(17∶83)为流动相;检测波长为316 nm;柱温35 ℃。
2.3.2对照品溶液的制备。取阿魏酸对照品11.44 mg,精密称定,置50 mL棕色量瓶中,加70%甲醇制成0.228 8 mg/mL的溶液,作为储备液。
2.3.3供试品溶液的制备。取本品20片,除去包衣,研细(过三号筛),取约2 g,精密称定,置具塞锥形瓶中,精密加入70%甲醇25 mL,密塞,称定重量,加热回流1 h,放冷,再称定重量,用70%甲醇补足减失的重量,摇匀,静置,取上清液滤过,取续滤液,即得。
2.3.4方法学考察。
2.3.4.1线性曲线的绘制。精密量取对照品储备液,分别制成2.265、4.530、6.795、9.060、18.121、22.651 μg/mL对照品溶液,以峰面积为纵坐标、浓度为横坐标绘制线性曲线,其线性方程为y=5E+07x-2 062.5(R2=0.999 9),表明阿魏酸在2.265~22.651 μg/mL线性关系良好。
2.3.4.2精密度、稳定性、重复性试验。取“2.3.2”项下的对照品溶液,按“2.3.1”色谱条件,连续进样6次,记录峰面积并计算其含量,得出阿魏酸含量的RSD为 0.4%,表明测定仪器精密度良好。取同一供试品溶液(批号190701),按照“2.3.1”色谱条件,分别于0、4、8、12、16、24 h进行测定,记录峰面积,计算得到阿魏酸含量的RSD为2.0%,说明供试品溶液在24 h内稳定。取同一批样品(批号190701)粉末2 g,精密称定,共称取6份,按“2.3.3”方法制备供试品溶液,并按“2.3.1”色谱条件进样测定,记录各样品的峰面积,计算得出阿魏酸含量的RSD为3.3%,表明该分析方法稳定,重复性良好。
2.3.4.3加样回收试验。取已知含量的样品(批号190701)粉末约1 g,精密称定,共称取6份,置具塞锥形瓶中,分别加入阿魏酸对照品溶液(0.018 12 mg/mL)4.0 mL,按“2.3.3”方法制备供试品溶液,并按“2.3.1”色谱条件进样测定,记录峰面积,计算得到阿魏酸回收率在95.66%~99.75%,平均回收率为97.38%,RSD为1.93%(表1),表明提取和检测方法准确可行。
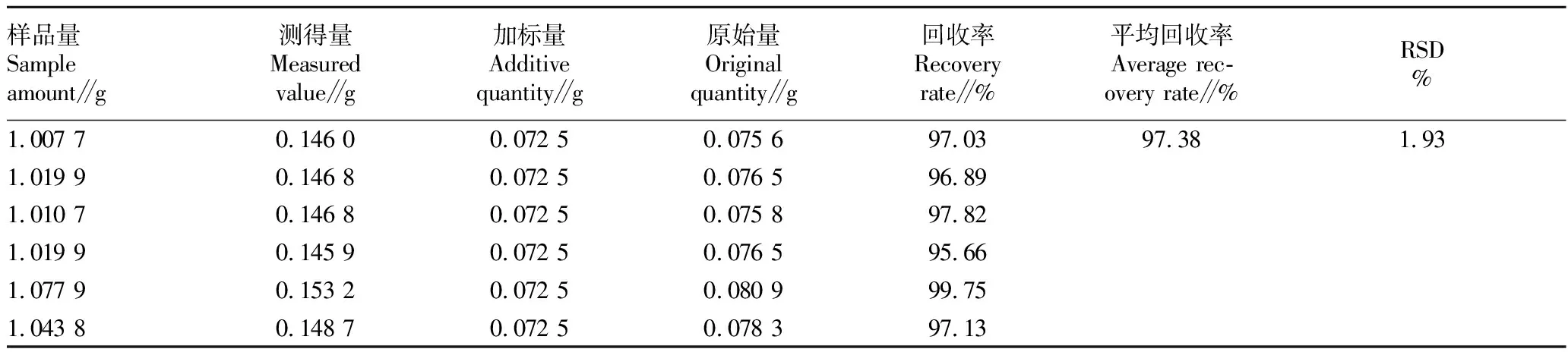
表1 阿魏酸加样回收率(n=6)
2.3.4.4不同仪器、不同品牌色谱柱考察。
(1)沃特斯液相色谱不同色谱柱比较。取样品(批号190701)2 g,精密称定,按“2.3.3”方法制备供试品溶液,按“2.3.1”色谱条件,以La Chrom C18(4.6 mm×250 mm,5 μm)、Agilent Eclipse Plus C18(4.6 mm×250 mm,5 μm)、Xbridge C18(4.6 mm×250 mm,5 μm)为色谱柱,进样测定,测得阿魏酸的含量分别为32.8、33.1、32.7 μg/片。因此不同品牌的色谱柱对阿魏酸的含量影响不明显。
(2)日立液相色谱不同色谱柱比较。取样品(批号190701)2 g,精密称定,按“2.3.3”方法制备供试品溶液,按“2.3.1”色谱条件,以La Chrom C18(4.6 mm×250 mm,5 μm)、Agilent Eclipse Plus C18(4.6 mm×250 mm,5 μm)、Xbridge C18(4.6 mm×250 mm,5 μm)为色谱柱,进样测定,测得阿魏酸的含量分别为32.9、33.0、32.8 μg/片。因此不同品牌的色谱柱对阿魏酸的含量影响不明显。
2.3.4.5当归原料药材考察。取当归原料粉末2 g,精密称定,按“2.3.3”方法制备供试品溶液,精密吸取供试品溶液10 μL,按“2.3.1”色谱条件注入高效液相色谱仪进行测定,测得当归原料药中阿魏酸含量为0.67 mg/g,其转移率为33.7%。
2.3.4.6提取时间的考察。取本品20片(批号191204),除去包衣,研细(过三号筛),取约2 g,精密称定,置具塞锥形瓶中,精密加入70%甲醇25 mL,密塞,称定重量,分别采用加热回流1、2、3 h时,放冷,再称定重量,用70%甲醇补足减失的重量,摇匀,静置,取上清液滤过,取续滤液,即得。测得阿魏酸含量分别为33.72、34.05、32.76 μg/片,阿魏酸含量差异不明显,因此为了考虑节省资源,选择提取时间为1 h。
2.3.5样品测定。分别取不同批号的加味归芪片、当归阴性粉末约2 g,精密称定,按“2.3.3”方法制备供试品溶液,按“2.3.1”色谱条件进样测定,具体结果见图4~5。从图4可以看出,加味归芪片中阿魏酸含量为29.6~34.0 mg/片,其中有一批阿魏酸(批号181101)的含量偏离。
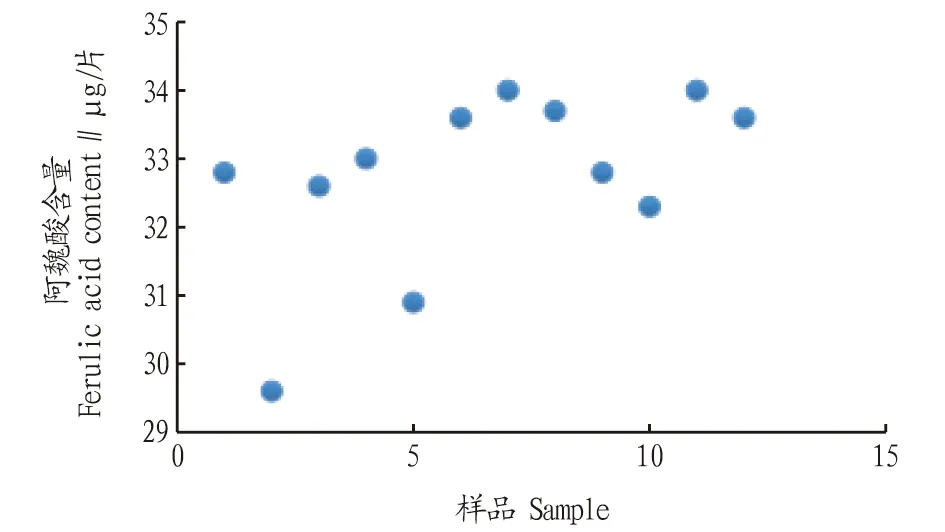
图4 加味归芪片中阿魏酸含量散点图Fig.4 Scatter plot of ferulic acid content in Jiawei Guiqi tablets
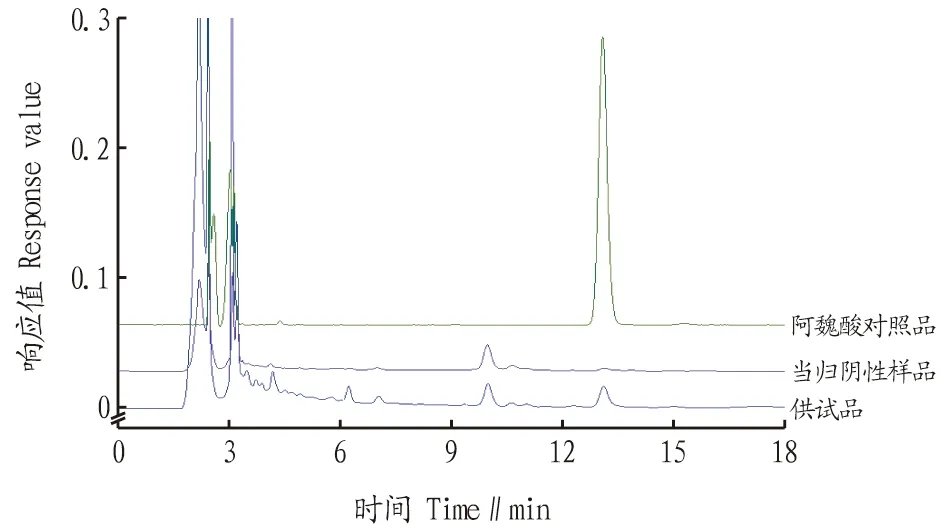
图5 加味归芪片中阿魏酸高效液相色谱图Fig.5 High performance liquid chromatography of ferulic acid in Jiawei Guiqi tablets
2.4 黄芪甲苷含量测定
2.4.1色谱条件。色谱柱为Agilent Eclipse Plus C18(4.6 mm×250 mm,5 μm);以乙腈-水(32∶68)为流动相;用蒸发光散射检测器检测。
2.4.2对照品溶液的制备。取黄芪甲苷对照品10.12 mg,精密称定,置25 mL棕色量瓶中,加甲醇制成0.404 8 mg/mL的溶液,作为储备液。
2.4.3供试品溶液的制备。取本品20片,除去包衣,研细(过三号筛),取约2 g,精密称定,加甲醇50 mL,密塞,超声处理30 min,放冷,再摇匀,滤过,洗滤纸数次,蒸干,残渣加水20 mL溶解,用水饱和的正丁醇振摇提取3次,每次40 mL,合并正丁醇液,用氨试液洗涤2次,每次40 mL,弃去氨液,正丁醇液蒸干,残渣加甲醇溶解,并转移至5 mL容量瓶中,加甲醇至刻度,摇匀,滤过,取续滤液,即得。
2.4.4方法学考察。
2.4.4.1线性曲线的绘制。精密量取对照品储备液,分别制成70.65、141.30、211.95、282.60、353.25、423.90、494.55 μg/mL 的对照品溶液,以1g峰面积为纵坐标、1g浓度为横坐标绘制线性曲线,其线性方程为y=1.782 7x+1.741 2(R2=0.999 8),表明黄芪甲苷在70.65~495.55 μg/mL线性关系良好。
2.4.4.2精密度、稳定性、重复性试验。取“2.4.2”项下的对照品溶液,按“2.4.1”色谱条件连续进样6次,测得峰面积,计算得出黄芪甲苷峰面积的RSD为0.1%,表明仪器精密度良好。精密吸取同一供试品溶液(批号190101),按“2.4.1”色谱条件分别于0、4、8、12、16、24 h进样测定,记录峰面积,计算得出黄芪甲苷峰面积的RSD为1.6%,表明该药品在24 h内稳定。取同一批样品(批号190101)粉末2 g,精密称定,共称取6份,按“2.4.3”方法制备供试品溶液,并按“2.4.1”色谱条件进样测定,记录各样品的峰面积,计算得出黄芪甲苷峰面积的RSD为2.3%,表明方法重复性良好。
2.4.4.3加样回收试验。取已知含量的样品(批号190101)粉末约1 g,精密称定,共称取6份,置具塞锥形瓶中,分别加入黄芪甲苷对照品溶液(1.248 181 mg/mL)1.0 mL,按“2.4.3”方法制备供试品溶液,按“2.4.1”色谱条件进样测定,记录峰面积,计算得到黄芪甲苷回收率在91.23%~97.74%,平均回收率为93.92%,RSD为2.59%(表2),表明提取和检测方法准确可行。
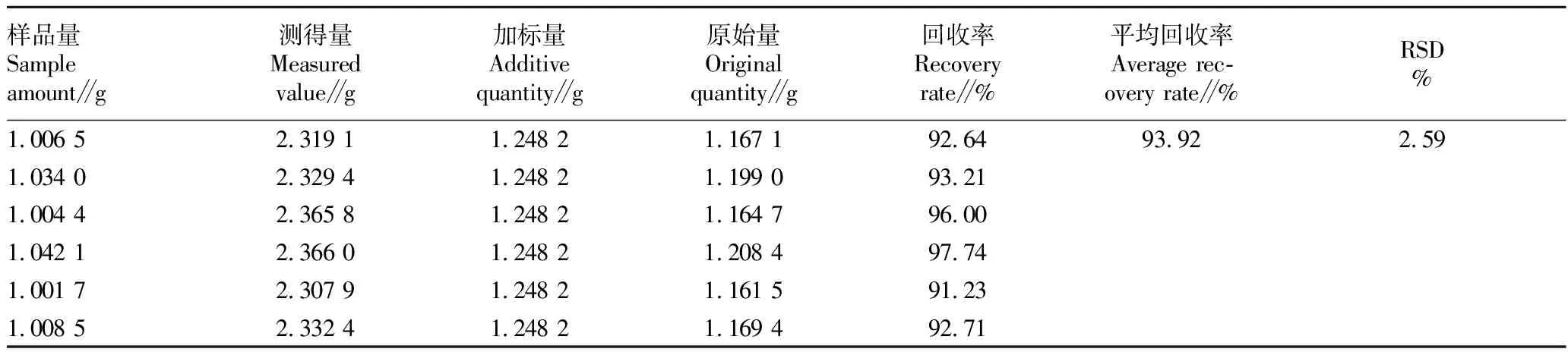
表2 黄芪甲苷加标回收率(n=6)
2.4.4.4不同品牌色谱柱考察。取样品粉末2 g(批号190101),精密称定,按“2.4.3”方法制备供试品溶液,按“2.4.1”色谱条件,以La Chrom C18(4.6 mm×250 mm,5 μm)、Agilent Eclipse Plus C18(4.6 mm×250 mm,5 μm)、Xbridge C18(4.6 mm×250 mm,5 μm)为色谱柱,进样测定,测得阿魏酸含量分别为0.408、0.410、0.420 mg/片。因此不同品牌的色谱柱对黄芪甲苷含量影响不明显。
2.4.4.5黄芪原料药材考察。取黄芪原料2 g,精密称定,按“2.4.3”方法制备供试品溶液,按“2.4.1”色谱条件进样测定,测得黄芪原料药中黄芪甲苷含量为0.917 mg/g,其转移率为79.7%。
2.4.4.6提取方法的考察。
(1)提取方法一。取3批样品(180501、190101、200104),分别取20片,除去包衣,研细(过三号筛),取约2 g,精密称定,加甲醇50 mL,密塞,超声处理30 min,放冷,再摇匀,滤过,洗滤纸数次,蒸干,残渣加水20 mL溶解,用水饱和的正丁醇振摇提取3次,每次40 mL,合并正丁醇液,用氨试液洗涤2次,每次40 mL,弃去氨液,正丁醇液蒸干,残渣加甲醇溶解,并转移至5 mL容量瓶中,加甲醇至刻度,摇匀,滤过,取续滤液,即得,测得3批样品180501、190101、200104中黄芪甲苷含量分别为0.421、0.420、0.428 mg/片。
(2)提取方法二。取3批样品(180501、190101、200104),分别取20片,除去包衣,研细(过三号筛),取约2 g,精密称定,置具塞锥形瓶中,精密加入含4%浓氨试液的80%甲醇溶液(取浓氨试液4 mL,加80%甲醇至100 mL,摇匀)50 mL,密塞,称定重量,加热回流1 h,放冷,再称定重量,用含4%浓氨试液的80%甲醇溶液补足减失的重量,摇匀,滤过,精密量取续滤液25 mL,蒸干,残渣用80%甲醇溶解,转移至5 mL容量瓶中,加80%甲醇至刻度,摇匀,滤过,取续滤液,即得,测得3批样品180501、190101、200104中黄芪甲苷含量分别为0.121、0.130、0.122 mg/片。
通过比较提取方法一和提取方法二可知,采用提取方法一测得黄芪甲苷含量明显高于提取方法二,因此该试验选择提取方法一。
2.4.5样品测定。分别取不同批号的加味归芪片、黄芪阴性粉末约2 g,精密称定,按“2.4.3”方法制备供试品,按“2.4.1”色谱条件进样测定,具体结果见图6~7。从图6可以看出,加味归芪片中黄芪甲苷含量为0.383~0.449 mg/片,其中有一批黄芪甲苷(批号181101)的含量偏离。
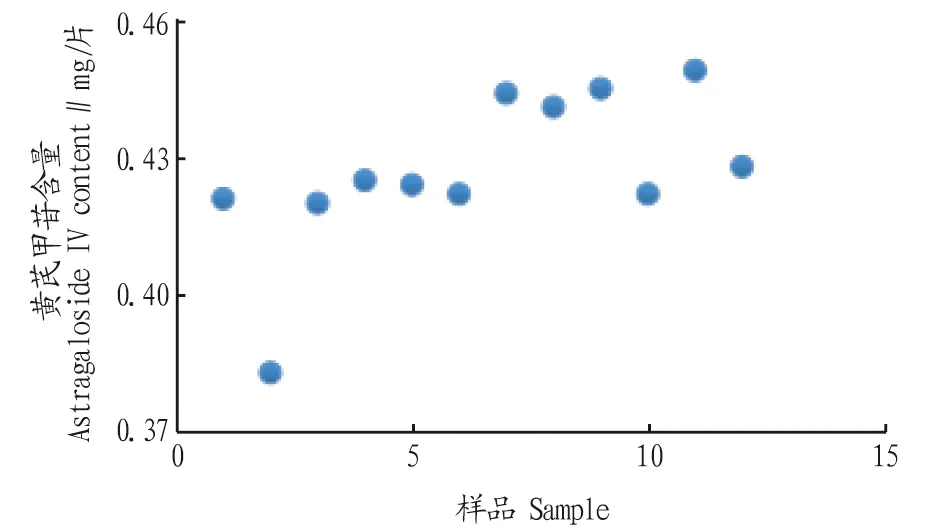
图6 加味归芪片中黄芪甲苷含量散点图Fig.6 Scatter plot of astragaloside IV content in Jiawei Guiqi tablets
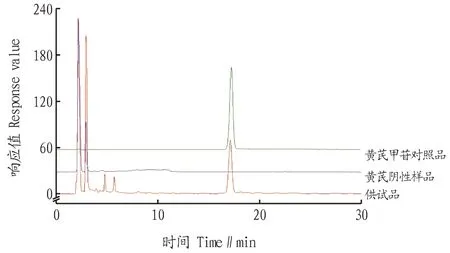
图7 加味归芪片中黄芪甲苷对照品高效液相色谱图Fig.7 High performance liquid chromatography of astragaloside IV reference substance in Jiawei Guiqi tablets
3 结论
加味归芪片组方分别为当归、黄芪、党参,该试验主要对该组方制剂中的当归、黄芪两味君臣药进行了研究。采用薄层色谱法,在不同温度不同湿度条件下考察了当归和黄芪的薄层色谱条件,结果发现在不同条件下,当归对照药材和阿魏酸对照品以及黄芪甲苷对照品在各自薄层条件下斑点清晰,分离效果明显,阴性均无干扰。因此该薄层色谱条件能很好地对加味归芪片中当归和黄芪进行定性鉴别。
加味归芪片中当归的含量考察采用阿魏酸指标成分,采用的高效液相色谱方法系统适应性符合要求,稳定性、精密度、重复性、回收率等方法学考察均能达到要求;采用不同仪器、不同品牌色谱柱等考察,系统适应性满足试验要求;同时考察不同提取时间对阿魏酸含量的影响,结果发现提取时间对阿魏酸含量影响不明显,为了经济又节省时间,因此选择提取时间为1 h。
加味归芪片中黄芪的含量考察采用黄芪甲苷指标成分,采用的高效液相色谱方法系统适应性符合要求,稳定性、精密度、重复性、回收率等方法学考察均能达到要求;考察不同品牌色谱柱,含量差异不明显,系统适应性满足试验要求;考察不同提取方法对黄芪甲苷的影响,结果测得含量差异明显,样品采用甲醇提取之后,再用水饱和的正丁醇溶液和氨试液分别萃取,得到的黄芪甲苷含量较高,因此选择该提取方法。
该研究建立的方法简单、方便、可行性和重复性良好,为加味归芪片质量标准提供技术支撑,同时更加有效控制加味归芪片的质量,确保了加味归芪片的疗效,为临床用药提供质量保证。
猜你喜欢
河南农业(2023年2期)2023-03-03
今日农业(2022年2期)2022-11-16
航天电子对抗(2022年4期)2022-10-24
今日农业(2021年6期)2021-06-09
特种经济动植物(2021年4期)2021-04-19
中成药(2018年7期)2018-08-04
中成药(2018年5期)2018-06-06
西部中医药(2015年9期)2015-02-02
安徽医药(2014年4期)2014-03-20
中成药(2014年8期)2014-02-28
