
Remdesivir,dexamethasone and angiotensin-converting enzyme inhibitors use and mortality outcomes in COVID-19 patients with concomitant troponin elevation
2023-10-17 13:23ChukwuemekaUmehHeatherMaozJessicaObiRuchiDakoriaSmitPatelGargiMaityPranavBarve
World Journal of Cardiology 2023年9期
Chukwuemeka A Umeh,Heather Maoz,Jessica Obi,Ruchi Dakoria,Smit Patel,Gargi Maity,Pranav Barve
Abstract BACKGROUND There are indications that viral myocarditis, demand ischemia, and renin-angiotensin-aldosterone system pathway activation play essential roles in troponin elevation in coronavirus disease 2019 (COVID-19) patients. Antiviral medications and steroids are used to treat viral myocarditis, but their effect in patients with elevated troponin, possibly from myocarditis, has not been studied.AIM To evaluate the effect of dexamethasone, remdesivir, and angiotensin-converting enzyme (ACE) inhibitors (ACEI) on mortality in COVID-19 patients with elevated troponin.METHODS Our retrospective observational study involved 1788 COVID-19 patients at seven hospitals in Southern California, United States. We did a backward selection Cox multivariate regression analysis to determine predictors of mortality in our study population. Additionally, we did a Kaplan Meier survival analysis in the subset of patients with elevated troponin, comparing survival in patients that received dexamethasone, remdesivir, and ACEI with those that did not.RESULTS The mean age was 66 years (range 20-110), troponin elevation was noted in 11.5% of the patients, and 29.9% expired. The patients' age [hazard ratio (HR) = 1.02, P < 0.001], intensive care unit admission (HR = 5.07, P < 0.001), and ventilator use (HR = 0.68, P = 0.02) were significantly associated with mortality. In the subset of patients with elevated troponin, there was no statistically significant difference in survival in those that received remdesivir (0.07), dexamethasone (P = 0.63), or ACEI (P = 0.8) and those that did not.CONCLUSION Although elevated troponin in COVID-19 patients has been associated with viral myocarditis and ACE II receptors, conventional viral myocarditis treatment, including antiviral and steroids, and ACEI did not show any effect on mortality in these patients.
Key Words: Coronavirus disease 2019;Troponin elevation;Remdesevir;Ace inhibitor;Steroids
INTRODUCTION
The coronavirus disease 2019 (COVID-19) pandemic caused by the severe acute respiratory syndrome coronavirus (SARS-CoV)-2 infection continues to have a devastating global impact, with over 640 million confirmed infections, including 6.6 million deaths worldwide as of December 3, 2022[1]. The virus's predilection for the pulmonary system is a well-studied effect of its pathogenicity, evidenced in the 2003 outbreak in Southern China. Cardiac involvement has also been a widely observed sequela of the COVID-19 infection, conferring a worse prognosis on those with underlying cardiovascular disease[2,3]. Several studies have discussed the usefulness of measuring cardiac troponins (cTn) as a measure of myocardial injury and also as a method to stratify at-risk individuals. Myocardial injury is defined as an elevation in cTn above the 99% of the upper reference limit (URL). It is considered acute if there is a subsequent rise and/or fall of cTn values[2,4].
Recent literature has revealed the cytopathic, inflammatory, and thrombotic effects of COVID-19 suggesting the important role of inflammatory markers in disease progression and severity[5,6] Particularly important is the correlation of the severity of the hyper-inflammatory response with a higher level of cTn, increasing the risk of mortality and complications in patients[2,7-9]. In addition, elevated C-reactive protein (CRP), procalcitonin, ferritin, D-dimer, interleukin (IL)-2, IL-7, granulocyte-colony stimulating factor, immunoglobulin G-induced protein 10, chemokine ligand 3 and tumor necrosis factor in COVID-19 patients, have been associated with cardiac injury and increased mortality[2,7-10].
Several mechanisms of myocardial injury leading to troponin elevation have been postulated. One such mechanism is direct virus-induced myocardial injury leading to myocarditis. In the initial phase, the virus facilitates its damage to the myocardium by direct lysis of cardiac myocytes[11]. This is typically followed by an intensified T-cell response, leading to further immune mediated myocyte injury and ventricular dysfunction[11]. Since 2005, it has been known that the SARSCoV virus infects cells through the angiotensin-converting enzyme (ACE) II receptor. ACE II receptors are highly expressed in vascular endothelium, cardiac pericytes, and alveolar cells, which has lent support to this proposed mechanism of myocardial damage[11,12]. This observation has now sparked interest in evaluating whether medications such as ACE inhibitors (ACEI) and angiotensin receptor blockers (ARBs) would benefit or harm those infected with SARS-CoV-2. In addition, SAR-CoV-2's indirect effects on the cardiovascular system have been studied extensively concerning ACE II downregulation leading to activation of the renin-angiotensin-aldosterone system (RAAS)[11]. Furthermore, ACE II catalyzes the conversion of angiotensin II to angiotensin I, a process that directly opposes the innate immune system's subsequent release of proinflammatory cytokines, vasoconstriction, pro-oxidants, pro-proliferative and profibrotic actions[11]. As such, there is interest in examining if inhibition of the ACE II enzyme with widely used antihypertensives, including ACEI and ARBs, would benefit COVID-19 patients as it stimulates the innate immune system, thus facilitating a more robust response to combat the underlying infection.
In summary, there are indications that viral myocarditis, demand ischemia, and RAAS pathway activation play essential roles in troponin elevation in COVID-19 patients. Antiviral medications and steroids are used to treat viral myocarditis, but their effect in patients with elevated troponin, possibly from myocarditis, has not been studied. Additionally, there is a lack of studies on the impact of ACEI use in patients with elevated troponin. Therefore, this multicenter retrospective study aims to evaluate the effect of dexamethasone, remdesivir, and ACEI on mortality in COVID-19 patients with elevated troponin.
MATERIALS AND METHODS
We conducted a multicenter retrospective observational study at seven hospitals in Southern California, United States. The study enrolled 1788 consecutive COVID-19 patients admitted to the seven hospitals between March 2020 and August 2021 who had a troponin test on admission. All patients were confirmed to have COVID-19 infection through a positive polymerase chain reaction nasopharyngeal swab. We extracted relevant deidentified patient data using a SQL program from the electronic medical record, which included: Age, race, gender, comorbidities, date of hospital admission, date of discharge, laboratory results on admission, medications they received while on admission, heart rate, and disposition at discharge. Our primary outcomes were the predictors of troponin elevation in COVID-19 patients and the effect of dexamethasone, remdesivir, and ACEI on mortality in COVID-19 patients with elevated troponin. Elevated cTn diagnostic of myocardial infarction is when the levels exceed the 99th percentile of a normal, healthy reference population (URL). This value is determined for each specific troponin assay with appropriate quality control in each laboratory[13]. Based on our laboratory assay, a troponin I (cTnI) level above 0.4 ng/mL was considered elevated for our study.
We performed a univariate analysis of the independent variables, including patients' age, gender, race, length of hospital stay, comorbidities, the medication patients received while in the hospital, and laboratory results using means and percentages. Furthermore, we performed a bivariate analysis of the relationship between troponin elevation and different study variables using chi-square andt-test, with aPvalue of 0.05 considered significant. We then did a backward selection logistic regression to determine the factors associated with troponin elevation. Additionally, we did a backward selection Cox multivariate regression analysis using mortality as a dependent variable. For the logistic and Cox regression analysis, we initially included statistically significant or biologically plausible variables from the bivariate analysis, such as patients' age, sex, body mass index, comorbidities, intensive care unit (ICU) admission, and mechanical ventilation, as independent variables in the multivariate model. The effect was expressed in odds and hazard ratios (HRs) for the logistic and Cox regression, respectively. Hypothesis testing was done using a two-sided test, and an alpha value of 0.05 indicated statistical significance.
In the second phase of our analysis, we did the Kaplan Meier survival analysis in the subset of patients with elevated troponin comparing survival in patients that received dexamethasone, remdesivir, and ACEI with those that did not. Statistical analysis was done using IBM SPSS version 27. The WIRB-Copernicus Group institutional review board approved the study.
RESULTS
Descriptive statistics
Tables 1 and 2 show the descriptive statistics of continuous and categorical variables of the study population, including length of stay, age, body mass index (BMI), and gender. The mean age was 66 years and ranged from 20-110 years, and 58% were males. Patients' race was categorized into white, Asian, black, and others, with 64.4%, 6.1%, 4.5%, and 24.9%, respectively. The patients had underlying comorbidities, including hypertension (42.3%), diabetes (19.2%), chronic kidney disease (CKD) (24%), and congestive heart failure (CHF) (18.8%). The patients received different medications while on admission, including dexamethasone (68.2%), remdesivir (50.9%), and ACEI (19.4%). Troponin was elevated in 11.5% of the study subjects and 29.9% of the total study population expired.
Bivariate analysis
In the bivariate analysis of continuous variables, length of hospital stay (P= 0.007), CRP (P< 0.001), lactate dehydrogenase (LDH) (P< 0.001), ferritin (P= 0.03), creatine phosphokinase (CPK) (P= 0.01), platelet count (P= 0.02), white blood cell count (P< 0.001), potassium (P< 0.001) and total bilirubin were significantly associated with troponin elevation. Patients with elevated troponin were more likely to have increased inflammatory markers, including CRP, LDH, ferritin, and CPK. The length of stay was also higher in patients with elevated troponin. There was no difference in age, BMI, and oxygen saturation in patients with or without elevated troponin (Table 3).
In the bivariate analysis of categorical variables, mortality (P< 0.001), ventilator use (P< 0.001), ICU admission (P< 0.001), CKD (P< 0.001), and CHF (P< 0.001) were significantly associated with elevated troponin. Patients with elevated troponin were more likely to die, be admitted to ICU, or be placed on a ventilator. Additionally, those with CHF or CKD were more likely to have elevated troponin (Table 4).
Multivariate analysis
In the multivariate logistic regression analysis, elevated levels of LDH [odd ratio (OR) = 1,P= 0.004], underlying CHF (OR = 2.7,P< 0.001), and ICU admission (OR = 3.6,P< 0.001) were independently associated with elevated troponin. Additionally, in the Cox regression multivariate analysis, age (HR = 1.02,P< 0.001), ICU admission (HR = 5.07,P< 0.001), and ventilator use (HR = 0.68,P= 0.02) were significantly associated with mortality. However, Troponin elevation (HR = 1.25,P= 0.1) was not independently associated with mortality after adjusting for age, comorbidities like CHF, ICU admission, and inflammatory markers (Tables 5 and 6).
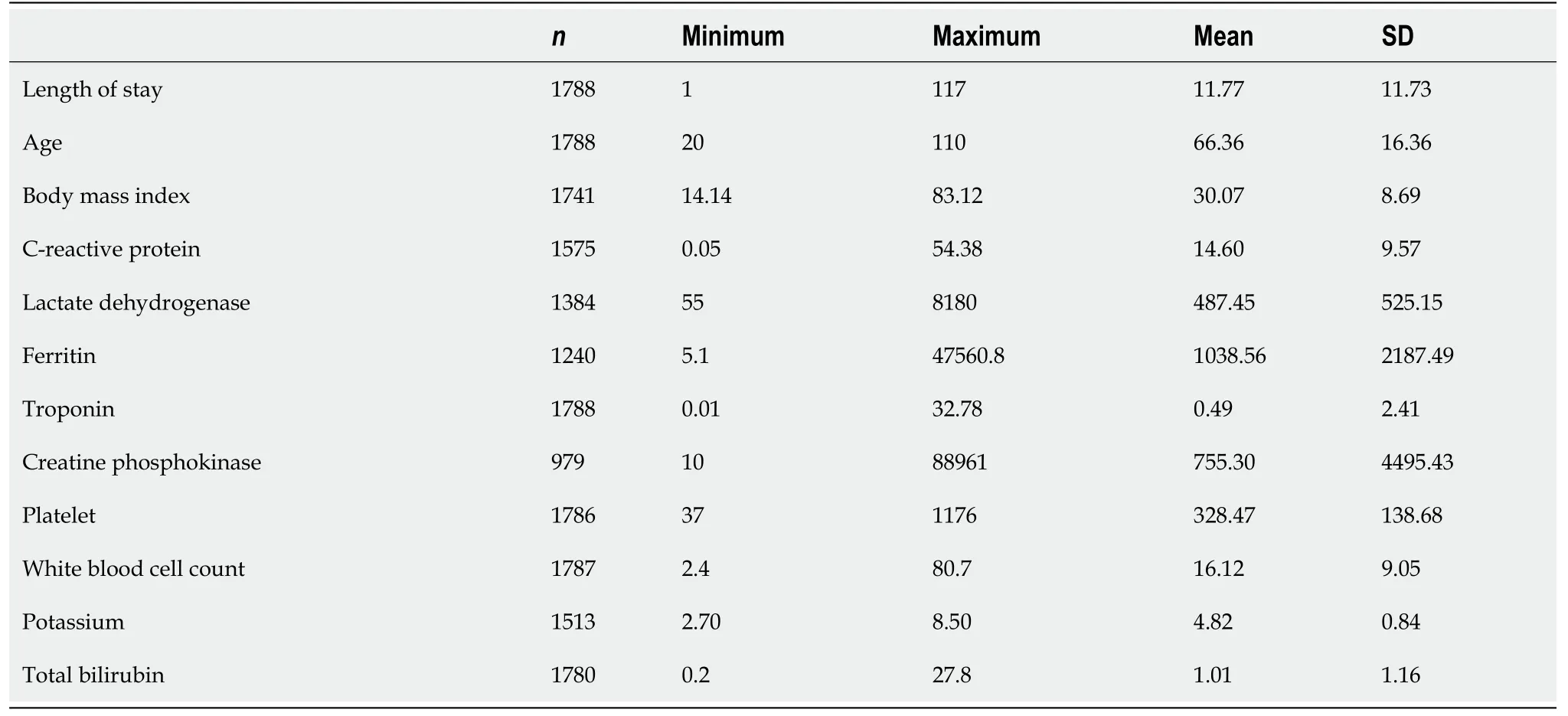
Table 1 Descriptive statistics of continuous variables
Subgroup analysis
In the Kaplan-Meier survival analysis of the subset of 205 patients with elevated troponin, the median survival in patients that received remdesivir (18 d) was higher than those that did not (14 d). However, this was not statistically significant (P= 0.07) (Figure 1A). Similarly, the median survival in patients receiving dexamethasone (16 d) was higher than in those not (14 d), but this was not statistically significant (P= 0.63) (Figure 1B). Finally, the median survival in patients on ACEI (16 d) was the same as those without (P= 0.8) (Figure 1C).
Furthermore, we did a Kaplan Meier analysis in the subset of patients with elevated concomitant troponin, CPK, and LDH, which would be expected in patients with significant myocarditis. However, similar to our analysis in those with elevated troponin, we found no difference in mortality between those that received dexamethasone (P= 0.88), ACEI (P= 0.83), or remdesivir (P= 0.93) and those that did not in this sub-group of patients.
DISCUSSION
COVID-19 has been reported to cause direct myocardial injury and myocarditis in some patients[14,15]. COVID-19 patients with myocarditis have increased troponin and non-specific ST-segment and T-wave changes. Echocardiogram typically shows global hypokinesis and pericardial effusion[14]. Thus, myocarditis mimics acute coronary syndrome, but the coronary arteries are usually normal on coronary angiogram. The gold standard for diagnosing myocarditis is histopathology of endomyocardial biopsy, an invasive procedure not often performed in clinical practice. Cardiac magnetic resonance imaging (MRI) is often used to diagnose myocarditis[14,16]. Antiviral medications, intravenous immunoglobulins, and immunosuppressants such as steroids and azathioprine have been used in treating viral myocarditis with limited evidence of benefit[16]. In our study, we did not do cardiac MRI to determine the proportion of patients with elevated troponin that have myocarditis. However, our Kaplan Meier analysis did not show any statistically significant difference in mortality in patients with elevated troponin who received steroids and those that did not. The role of steroids in myocarditis caused by COVID-19 is unclear, and the lack of steroid efficacy on mortality in patients with elevated troponin in our study could suggest that steroids may not play any significant role in COVID-19 induced myocarditis. However, we do not know the proportion of patients in our study with myocarditis, and the role of steroids in COVID-19 myocarditis needs to be further investigated.
Furthermore, studies have suggested a possible relationship between increased viral load and myocardial injury in COVID-19 patients. For example, in a study of hospitalized COVID-19 patients, those with detectable viremia were significantly more likely to have elevated troponin and myocardial injury than those without viremia[17]. Although another study did not find any relationship between the initial viral load and the incidence of myocardial injury in hospitalized COVID-19 patients, high viral load and myocardial injury were independent predictors of in-hospital mortality[18]. Remdesivir, a viral RNA polymerase inhibitor, significantly reduced the median recovery time of COVID-19 patients compared to placebo in the adaptive, randomized controlled Adaptive COVID-19 Treatment Trial-1 study[19]. However, there is limited data on how Remdesivir impacts cardiac injury. In the Kaplan-Meier survival analysis of the subset of patients with elevated troponin in our study, the median survival in patients that received remdesivir (18 d) was higher than those that did not (14 d). However, this was not statistically significant (P= 0.07). The lack of statistical significance could be due to our sub-group analysis being underpowered to detect a difference.
SARS-CoV-2 binds to the ACE II receptor (ACE II), which is highly expressed in the lungs and myocardium, and this has been postulated as a mechanism through which the virus causes direct damage to cardiac cells[20]. ACEI have been shown to upregulate the expression of ACE II in lung cells in animal studies. The mechanism is unclear but is possiblythrough decreasing angiotensin II, leading to indirect upregulation of ACE II[21,22]. Thus, there have been concerns that the use of ACEI in patients with COVID-19 will increase the risk of lung and myocardial injury. However, some human studies did not support the hypothesis that ACEI use increases ACE II expression and the risk of lung and myocardial injuries in COVID-19 patients[23]. Furthermore, the Kaplan-Meier analysis in our study showed no difference in survival in patients with elevated troponin who received ACEI and those who did not, suggesting that the use of ACEI should not be withheld even in patients with elevated troponin.
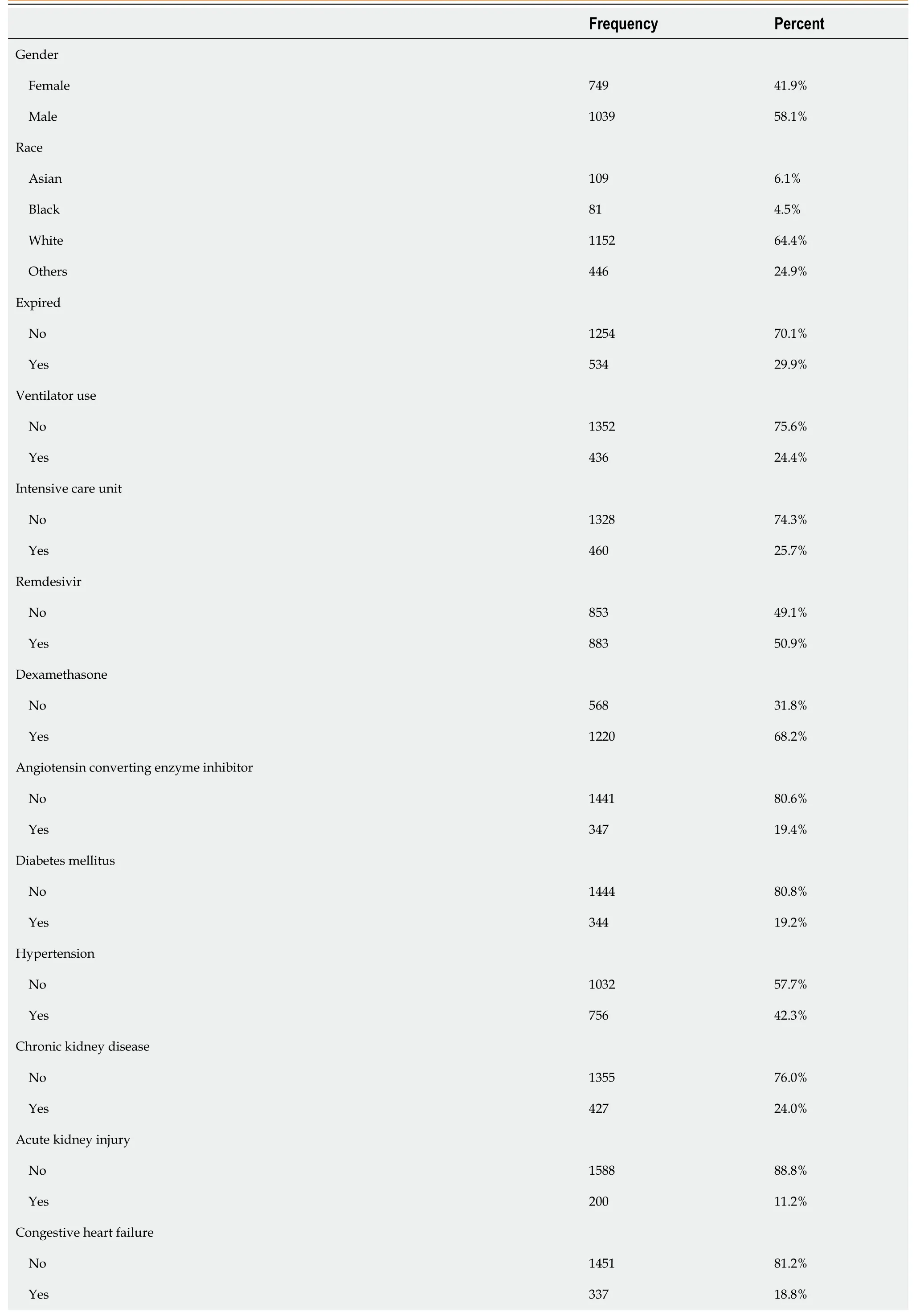
Table 2 Descriptive analysis of categorical variable
In our study, elevated levels of LDH (OR = 1,P= 0.004), underlying CHF (OR = 2.7,P< 0.001), and ICU admission (OR = 3.6,P< 0.001) were independently associated with elevated troponin. This finding is similar to former studies that showed that elevated troponin, increased age, and co-morbidities are predictors of ICU admission[24]. This was further collaborated by a meta-analysis of 23 studies that showed that patients with elevated troponin had a significantly increased risk of severe disease and ICU admission [risk ratio (RR) = 5.57, 95%confidence interval (95%CI): 3.04 to 10.22,P< 0.001; RR = 6.20, 95%CI: 2.52 to 15.29,P< 0.001][25]. Furthermore, our study showed that elevated troponin was associated with CHF, similar to previous studies that showed that patients with troponin elevation were older, with more co-morbidities[26].
In our study, there was increased mortality in patients with troponin elevation (HR = 1.25,P= 0.1), although this was not statistically significant at aPvalue of 0.05. The lack of statistical significance could be related to the fact that our study was underpowered to detect a difference. It could also be that patients in our study differ from those in previous studies. For example, while the proportion of patients in our study with elevated troponin was 11%, a meta-analysis of prior studies has shown an average of 31% (range 23%-38%)[27], 22.9%[28], and 27% (range 9%-51%)[29]. Studies have found that elevation of high-sensitivity troponin and traditional troponin assays are associated with increased mortality in COVID-19 patients[27-31]. Patients with elevated troponin had significantly increased odds of death than those with normal troponin independent of elevation in inflammatory markers and cardiovascular co-morbidities[27-31].
Our study has several limitations. Firstly, in the subgroup analysis, we dealt with a small sample size which may limit the overall power of the study. Secondly, troponin was assessed on admission and was not monitored for the duration of the patient’s hospital stay, which may impact the lack of association observed between troponin elevation and mortality. Thirdly, in the analysis between ACEI use and troponin elevation, our data only reflects patients placed on an ACEI during their hospital stay. We did not stratify patients on whether they were on the medication previously, and it is possible some patients on ACEI at home might not have been started on it in the hospital. This might have resulted in a misclassification bias and affected the study outcome. Finally, this research is an observational study, and there might have been unmeasurable variables that might have confounded the study outcome. Also we did not conduct cardiac MRI to confirm out findings of myocarditis but based that on inferential analysis.
CONCLUSION
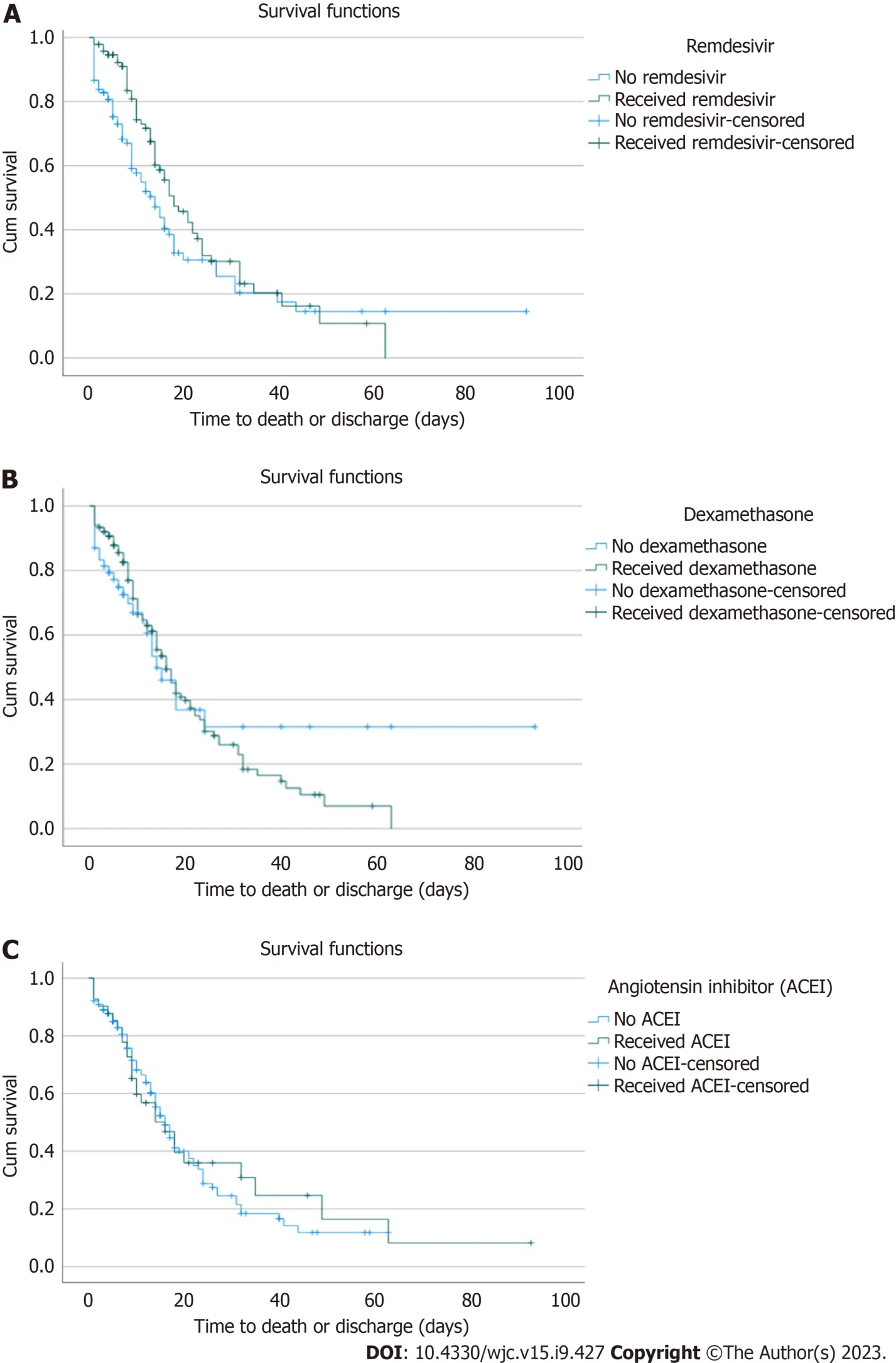
Figure 1 Kaplan Meier.A: Remdesivir use and mortality in patients with troponin elevation;B: Dexamethasone use and mortality in patients with troponin elevation;C: Angiotensin-converting enzyme inhibitors use and mortality in patients with troponin elevation.ACEI: Angiotensin-converting enzyme inhibitors.
Although elevated troponin in COVID-19 patients has been associated with viral myocarditis, conventional viral myocarditis treatment, including steroids and antiviral, did not affect mortality in these patients. In addition, previous studies have suggested a possible association between COVID-19 viral load and myocardial injury; however, we found no statistically significant difference in survival in patients with elevated troponin treated with remdesivir and those that were not. Furthermore, our study suggested that ACEI should not be withheld even in patients with elevated troponin because it did not negatively or positively affect survival.
ARTICLE HIGHLIGHTS
Research background
Several studies have proposed that troponin elevation seen in coronavirus disease 2019 (COVID-19) patients is due to an interplay between viral myocarditis, demand ischemia and renin-angiotensin-aldosterone system pathway activation. This creates the hypothesis that the use of steroids, antivirals and angiotensin-converting enzyme inhibitors (ACEI) in patients with COVID-19 infection and troponin elevation would impact mortality outcomes.
Research motivation
The COVID-19 pandemic has had a monumental global impact and resulted in several deaths worldwide. The motivation of this study was to analyze if the use of the steroids, antivirals and ACEI would improve survival in patient with COVID-19 infection and troponin elevation.
Research objectives
Our main objective was to analyze any differences in mortality in our subjects, in the hopes of adding to existing knowledge and creating a standardized treatment protocol in patients with COVID-19 and troponin elevation.
Research methods
Our study design was a retrospective observational study consisting of 1788 COVID-19 patients at seven hospitals across Southern California. To determine the predictors of mortality in our subjects, we did a backward selection cox multivariate regression analysis. Furthermore, to analyze survival in the subset of patients with troponin elevation we did a Kaplan Meier analysis comparing those that received treatment with steroids, remdesivir and ACEI and those that did not.
Research results
Though the beneficial role of steroids in the treatment of COVID-19 has been established, our study did not show any statistically significant difference in mortality in patients with elevated troponin who received steroids and those that did not. Therefore, the role of steroids in myocarditis caused by COVID-19 is still unclear and needs further investigation. On the other hand, our study showed improved survival in COVID-19 patients with elevated troponin that received remdesivir, although this was not statistically significant.
Research conclusions
Although the mechanism of troponin elevation in COVID-19 patient has been linked to viral myocarditis and reninangiotensin-aldosterone system activation, the novel treatments of these subsequent pathologies including steroids, remdesivir and ACEI showed no significant survival benefit in our study. This creates the theory that there are other mechanisms at play guiding this complex interaction.
Research perspectives
Although our study did not show a statistically significant mortality benefit with the use of steroids and remdesivir, our sub-group analysis was limited by a small sample size, so further studies on the effect of remdesivir in the sub-set of COVID-19 patients with elevated troponin using a larger population will be beneficial.
FOOTNOTES
Author contributions:Umeh CA, Maoz H, Obi J, Dakoria R, Patel S, Maity G and Barve P conceptualized and revised the study design; Umeh CA analyzed the data; Maoz H, Umeh CA, Obi J, Dakoria R, Patel S, and Maity G, wrote the first draft of the paper; Barve P and Umeh CA, reviewed and revised the paper; Maoz H led and coordinated the research and writing of the manuscript; Barve P and Umeh CA supervised the project; all authors have read and approved the final manuscript.
Institutional review board statement:The WIRB-Copernicus Group (WCG) institutional review board (IRB) approved the study. IRB approval number: 13410516.
Informed consent statement:Our study was a retrospective observational study which used medical records for data acquisition and analysis and thus does not require informed consent from subjects.
Conflict-of-interest statement:None to declare.
Data sharing statement:The data supporting the findings of this study are available from the corresponding author upon reasonable request.
Open-Access:This article is an open-access article that was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution NonCommercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: https://creativecommons.org/Licenses/by-nc/4.0/
Country/Territory of origin:United States
ORCID number:Chukwuemeka A Umeh 0000-0001-6574-8595;Heather Maoz 0009-0000-5913-9902;Jessica Obi 0009-0000-7812-591X;Ruchi Dakoria 0009-0006-9131-919X;Smit Patel 0009-0007-1659-8018;Gargi Maity 0009-0008-9758-9638;Pranav Barve 0000-0002-3490-1451.
S-Editor:Lin C
L-Editor:A
P-Editor:Xu ZH
 World Journal of Cardiology2023年9期
World Journal of Cardiology2023年9期
- World Journal of Cardiology的其它文章
- Real-time cardiovascular magnetic resonance-guided radiofrequency ablation: A comprehensive review
- Immediate in-hospital outcomes after percutaneous revascularization of acute myocardial infarction complicated by cardiogenic shock
- Outcomes in patients with COVID-19 and new onset heart blocks:Insight from the National Inpatient Sample database
- Variant of Wellen’s syndrome in type 1 diabetic patient: A case report