
六氟化硫替代气体的研究现状及未来发展趋势
2023-10-07 12:34杨志强曾纪珺马义丁尉涛赵波刘英哲张伟吕剑李兴文张博雅唐念李丽孙东伟
化工进展 2023年8期
杨志强,曾纪珺,马义丁,尉涛,赵波,刘英哲,张伟,吕剑,李兴文,张博雅,唐念,李丽,孙东伟
(1 西安近代化学研究所氟氮化工资源高效开发与利用国家重点实验室,陕西 西安 710065;2 西安交通大学电力设备电气绝缘国家重点实验室,陕西 西安 710049;3 广东电网有限责任公司电力科学研究院,广东 广州 510699)
特高压电力输送是建立我国新型多能源电力通道,实现大容量长距离“西电东送南电北送”的关键环节。气体绝缘设备是特高压电网的重要组成部分[1],六氟化硫(SF6)以其优异的绝缘与灭弧性能广泛应用于输电装备,是绝缘设备中最重要的气体绝缘和灭弧介质[2-3]。每年约有8000t SF6气体应用于电力工业领域[4]。然而,SF6的温室效应是CO2的23900 倍[2],《京都议定书》《巴黎协定》等国际公约中明确限制了该气体的排放和使用[4]。
为应对气候变化,我国提出了2030年碳达峰、2060 年碳中和的“双碳”目标。在全球减碳的背景下,采用兼顾电气与环境性能的环保绝缘气体是电网装备发展的必然趋势。目前主要研究的SF6替代气体为八氟环丁烷(c-C4F8)、三氟碘甲烷(CF3I)、氢氟烃(HFCs)、氢氟烯烃(HFOs)、全氟异丁腈(C4F7N)、全氟戊酮(C5F10O)、三氟硫氮(NSF3)等。然而,上述气体难以兼顾绝缘、液化、环保和安全等性能,探索新型环保绝缘气体成为近年来国内外研究的热点。
本文以环保型SF6替代气体的性能需求为出发点,回顾了SF6替代气体的探索历程,指出环保绝缘气体各项宏观性能的固有矛盾,传统气体筛选方法难以创造出兼顾各项性能的新型气体。针对目前的问题与挑战,总结了现阶段绝缘性能和制备方法的研究成果,随后对SF6替代气体构效关系模型与分子设计的优势及适用性进行了综述,进而提出了高通量分子设计与共沸气体的替代气体筛选方案,旨在为我国SF6替代气体的探索提供一定的参考与借鉴。
1 SF6替代气体的筛选方法
国内外对SF6替代气体的筛选策略、构效关系、分子设计等方面进行了广泛的研究[5-8],理想的环保绝缘气体应满足绝缘性能高、不易液化、温室效应潜值(GWP)低、不破坏臭氧层、稳定性高、安全无毒等多项指标要求[5]。
绝缘气体为电负性气体,其绝缘性能来源于气体分子对电子的强捕获能力,能够极大减缓负离子的运动速度,从而抑制放电过程中电子崩的发生[9-11]。其绝缘强度通常用相对于SF6气体的绝缘强度Er(量纲为1)来表征。GWP是用于表征气体环保性能的重要指标,是相对于二氧化碳温室效应的比值。从分子结构的角度来看,绝缘强度与液化温度、分子稳定性与温室效应之间相互制约(图1),单一气体的宏观性质存在难以克服的固有矛盾。气体分子的尺寸越大,捕获电子的截面积越大,有利于提高绝缘强度,但是分子尺寸的增加,同时会降低分子自身的运动能力,又会导致液化温度升高;分子极性增强,分子的偶极矩增加,有利于捕获电子,提高绝缘强度,但是极性分子间的取向力和诱导力都增强,使得分子间更倾向于凝聚,同样会导致液化温度升高[12-13]。除此之外,绝缘气体还需要考虑稳定性和环保性能,稳定性高的气体往往与对流层羟基自由基反应速率慢,难以自然降解,因此具有较高的GWP,温室效应明显[14-15]。

图1 单质气体宏观性的固有矛盾
绝缘气体的分子设计和筛选需要在环保性能、毒性、液化温度、绝缘性能之间进行权衡[5,13]。就已知的上亿种化合物而言,能够满足高绝缘强度的分子众多,但同时满足高绝缘强度和低液化温度的分子数量则呈指数减少,每增加一项筛选要求,可选分子数量呈指数削减。因此,环保绝缘气体的研制极具挑战性,完全满足所有指标要求的单质气体至今尚未发现[16]。如何通过揭示绝缘、液化、环保等气体特性的物理化学本质,突破新型绝缘气体的构建策略是研发的核心问题。目前,绝缘气体的筛选方法包括试错法和计算机辅助设计方法。
1.1 试错法
试错法是科研人员根据以往经验筛选出样品,结合分析测试结果评估样品性能是否满足需求。如不满足指标,则更换目标化合物,直到样品的性能满足要求。传统的绝缘气体筛选是以试错法为主,科研人员根据经验在已知化合物数据库中进行多项性能评估,逐一进行气体性能的测试和筛选,寻找满足要求的SF6替代气体。20世纪80年代,美国西屋电气公司系统地开展了绝缘气体的筛选工作,考察了近400种气体的绝缘、毒性、理化等性质,筛选出约39种沸点低于-10℃的电负性气体和28种沸点高于-10℃的添加物,未找到一种气体能够全面优于SF6[17]。Devins[18]系统测量了35种气体的击穿电压与部分沸点,研究了分子结构对绝缘性能的影响,认为能够真正替代SF6的分子结构数量有限,以含Cl、CN、C≡C结构为主。但是,电气实验昂贵且非常耗时,仅依靠试错实验很难实现结构-性能的调控,通过试错法寻找替代SF6的环保气体极其困难。
1.2 计算机辅助设计法
随着高性能计算技术发展和计算化学方法工具化,计算机辅助设计方法已经被广泛应用于药学、材料科学、化学等多学科领域。相比于试错法,计算机辅助设计可有的放矢地进行结构筛选,大大降低了人工和实验成本。通过计算机开展分子结构设计和性能预估,从分子拓扑结构、电子结构和需求性能之间的关系出发,设计同时满足绝缘强度高、沸点低以及GWP 低等条件的气体分子,从分子结构层面上解决各理化性质之间难以平衡的突出问题,已成为当前SF6替代气体筛选的主流研究趋势。
计算机辅助设计能够预测未知化合物的性能,从而对基团取代后的新化合物的进行性能预估。张超海等[19]通过—F、—Cl、—CH3、—CF3、—NH2、—NF2、—CN 基团取代SF6中氟原子,研究了取代后气体绝缘强度和沸点的变化规律。研究表明—Cl、—NF2和—CN基团取代可以提高气体绝缘性能,但是气体其他性能如沸点、化学稳定性以及生物毒性难以满足要求。余小娟等[6]通过—CH3、—NF2、—Cl 以及—CN 基团取代SF6中氟原子,结合构效关系模型分析了取代气体绝缘强度和液化温度的变化规律。王宝山等[20]采用构效关系模型预测了已知化合物SF3N 绝缘强度和液化温度,其中—S≡N 取代了SF6中的—SF3,该分子的绝缘强度是SF6的1.3 倍,液化温度-27℃,不饱和键的引入使其GWP 远低于SF6。余小娟等[21]以SF6、c-C4F8、CF4、C2F6分子为骨架采用N2、C2F4、C2F2、CO2、SO2分子片段进行官能团取代,形成了12种新型绝缘气,通过计算机辅助预测了其多种性能,获得了3种潜在SF6替代物(F3SN、F4SCF2、F3SCF)。
从已知分子数据库出发,设置GWP、绝缘性能、液化温度等边界条件,同时结合构效关系模型对分子进行筛选,是另一种研究思路。Kazakov等[13]从PubChem 数据库的56203 个分子中进行结构筛选,依次采用GWP、燃烧下限(L)、临界温度(Tc)、毒性和化学稳定性5 个限定条件筛选出了1234 个制冷剂候选物。Rabie 等[5]在此基础上通过引入绝缘强度(Er)和沸点(Tb)2 个维度的构效关系,开展了SF6替代气体筛选研究,筛选出7 种SF6潜在替代物。
计算辅助设计的研究进展表明:以SF6为骨架的分子设计高度依赖经验,基团取代使分子的种类和数量受到很大的局限,未知新型结构的分子设计研究还有待开展;数据库分子数不足,预测模型不精确,影响了筛选效果,如何构建更精准的绝缘、液化、GWP 的预测模型,同时扩大分子筛选数据库,是计算机辅助筛选未来的主要发展方向。
2 潜在SF6替代气体及其合成方法
20 世纪90 年代后期,由于温室效应问题的加剧,SF6替代气体的研究开启了新的篇章。人们开始尝试用新的环保气体替代SF6,全氟异丁腈(C4F7N)[22]、全氟戊酮(C5F10O)、三氟硫氮(NSF3)等新型气体因其优异的绝缘性能和相对较低的GWP而受到关注(表1)。

表1 常见绝缘气体的基本性质
2.1 全氟酮
2014 年,瑞士ABB 公司提出了以C5F10O(C5-PFK)及C6F12O(C6-PFK)为代表的全氟酮类绝缘气体见图2。该类气体无毒、不可燃、GWP值约为1、ODP为0,绝缘强度为SF6气体的两倍,具有良好的绝缘和环保性能。然而,该类气体的液化温度较高(C5和C6纯气体的常压沸点为25℃和49℃),需要添加空气、CO2形成混合气体使用,ABB公司的击穿实验显示C5和C6混合气体的绝缘性能与SF6相当[23],具备代替SF6的潜力。
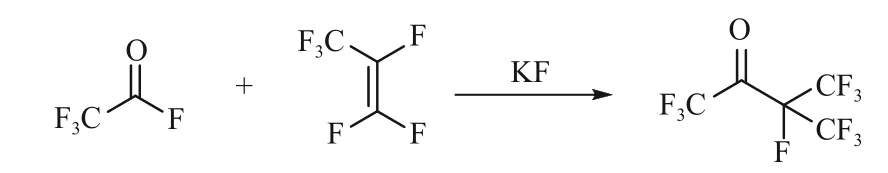
图2 三氟乙酰氟、六氟丙烯加成制备C5-PFK
全氟戊酮(C5-PFK)的主要合成方法是以六氟丙烯与三氟乙酰氟为原料通过加成反应制备,不仅反应步骤短且原子利用率较高。Coffman 等[24]首次以三氟乙酰氟与六氟丙烯加成反应制备获得了C5-PFK,原料转化率75%。2011 年,苏威公司的Wlassics 等[25]又以三氟乙酰氟和六氟丙烯为原料制备C5-PFK 的方法,通过调整反应溶剂、温度、操作工艺等,蒸馏获得了C5,收率70%。为改善反应氟盐后处理问题,Moldavskii 等[26]采用负载型的氟化铯,在无溶剂条件下,制备获得了C5,将收率提高至92%,后处理过程也得到了简化。
全氟己酮C6F12O(C6-PFK)制备方法与C5-PFK相似,Rivers 等[27]以全氟丙酰氟为原料,在金属氟化物活化下,与六氟丙烯加成,可获得C6-PFK,见图3。由于全氟丙酰氟大规模制备存在难度,因此改良工艺[28]以六氟环氧丙烷为原料,首先在氟化物作用下异构化为全氟丙酰氟,收率可达97%以上,再将异构化获得的全氟丙酰氟与六氟丙烯反应制备C6-PFK。
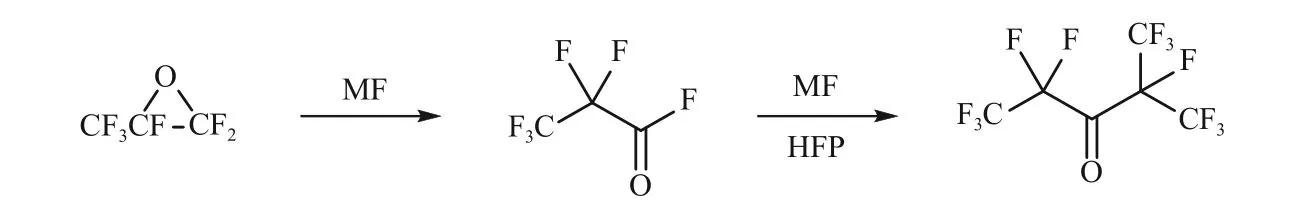
图3 C6-PFK的制备方法
2.2 全氟异丁腈
法国阿尔斯通公司也提出了新型环保气体全氟异丁腈(C4F7N、C4-PFN),该气体需要与CO2混合使用,纯C4-PFN 气体的GWP 为2100,ODP 为0,液化温度为-4.7℃,绝缘强度为SF6的2.2倍,制备方法见图4。近年来,C4-PFN 气体受到广泛的关注,Nechmi 等[29]采用交流和脉冲电压测量了C4-PFN/CO2混合气在不同电极条件下的击穿电压,结果表明C4F7N 与CO2具有很好的协同作用,C4-PFN的体积分数为3.7%时协同作用最强;3M 公司Kieffel 等[30]采用145kV GIS 样机测试了C4-PFN/CO2混合气体的绝缘强度,发现C4-PFN 含量为18%~20%时,混合气体具有与SF6相同的绝缘强度。

图4 C4-PFN的制备方法
Coffman 等[31]以碳酰氟和六氟丙烯为原料,在氟化钾催化下,首先获得了全氟异丁酰胺;在此基础上,武汉大学罗运柏教授课题组[32]以草酰氯和六氟丙烯为原料,通过亲核加成、胺解、脱水三步制备获得了C4-PFN,总收率42%。该方法是目前C4-PFN 的主要工业方案,但使用化学量的KF 和大量的冠醚,造成制备工艺不仅成本高,且难以连续化进行。Oxenrider等[33]采用六氟丙烯与乙二腈为原料制备得到了C4-PFN,室温搅拌反应4 天,全氟异丁腈粗品收率64.3%。为寻求更绿色的C4-PFN 制备方法,3M 公司采用电化学方法[34],以异丁酸酐与无水氟化氢为原料,通过电解氟化反应制备了C4-PFN,但由于电解过程复杂,副反应多,产物纯度难以满足电力应用企业的要求。
GE 公司提出了以C4-PFN 为核心介质的Green Gas For Gird(3气体)概念,并研制了相应的气体绝缘管道、气体绝缘开关和电流互感器等产品。C4-PFN 的缺点是温室效应指数较高,且生物毒性存在争议[35]。
2.3 三氟硫氮
1965年,美国联合化学公司[36]首次将三氟硫氮(NSF3)应用于绝缘气领域,测得NSF3气体的液化温度为-27.1℃,测量了NSF3与SF6混合物的绝缘强度变化,发现当NSF3浓度低于80%时,电击穿引起的NSF3降解就可以避免,NSF3与SF6混合气体能够提高SF6的绝缘强度。2020年,武汉大学再次测试了NSF3的物理和环境特性,报道了NSF3的GWP为916,绝缘强度为1.35,通过与主流SF6替代物对比,指出NSF3具有相对优异的综合性能,具有潜在的应用前景。
1955年,Glemser等[37]首次以S4N4与AgF2为原料制备获得了NSF3。2020年,罗运柏课题组[38]以二氯化二硫与液氨为原料,通过胺解、氟化两步反应,再次制备了NSF3,全程收率25%。遗憾的是,该方法副反应多,NSF3的最高纯度仅为90%左右,AgF2的摩尔当量高达10倍以上,导致该工艺难以放大。Clifford等[39]以氟代甲酰亚胺二氟化硫(F2S= = NCOF)与AgF2为原料,合成得到了NSF3,但AgF2的用量仍为近4倍当量。NSF3的制备工艺复杂,需使用昂贵的氟化试剂,迄今为止,合成的最大规模也不过克级,通过催化氟化等技术,开发NSF3新型制备工艺,可能是未来的研究重点。NSF3的主要制备方法见图5。
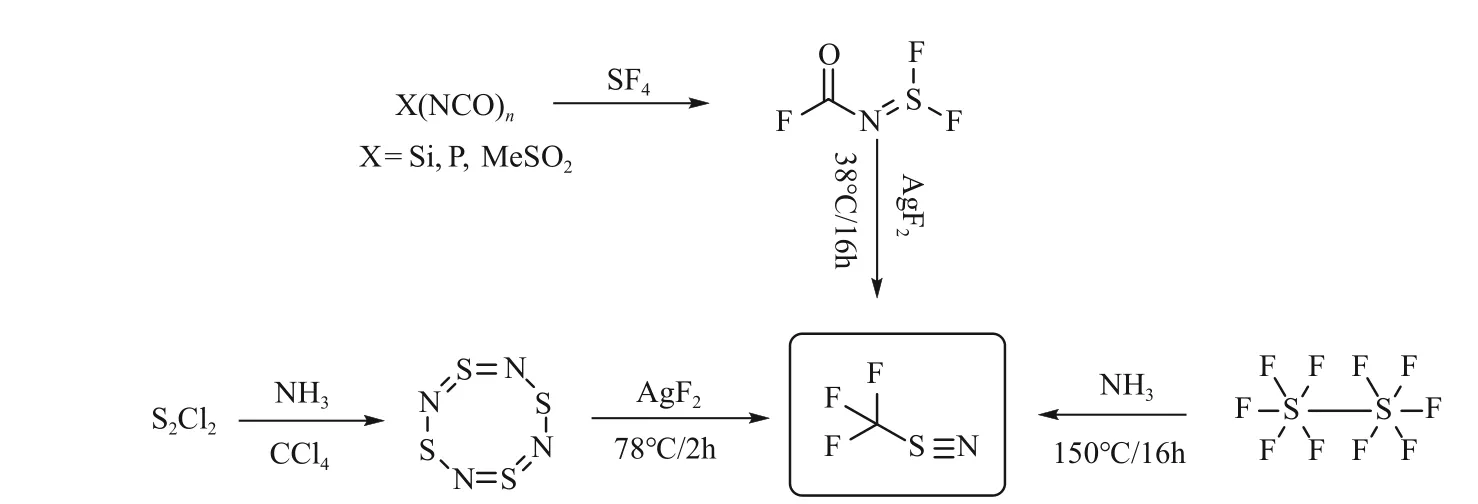
图5 NSF3的主要制备方法
2.4 氢氟烯烃
氢氟烯烃(HFOs)具有极低的GWP,且ODP为零、毒性低、不易燃,是一类综合性能优异的物质[16],被认为是理想的制冷剂替代品。近年来,国内外很多学者提出了以HFOs作为新型环保绝缘气体,主要制备方法见图6。法国施耐德公司从绝缘性能、材料相容性、毒性、液化温度、老化性能等多方面评估了HFO-1234ze(E)气体代替SF6的可行性[40],结果表明HFO-1234ze(E)具有良好的绝缘性能(约为SF6的83%)和最优的毒理测试结果,但灭弧能力较弱。苏黎世联邦理工学院Frank 等[41-42]采用脉冲汤逊法测量了HFO-1234ze(E)、HFO-1225ye(Z)及其混合气体的绝缘性能,获得了HFO-1234ze(E)、HFO-1225ye(Z)纯气体的电子群参数,确定了临界击穿场强,并发现了HFO1234ze(E)与N2、SF6气体的协同作用[8,43]。

图6 HFO-1234ze(E)的主要制备方法
反式-1,3,3,3-四氟丙烯[HFO-1234ze(E)]采用1,1,1,3,3-五氟丙烷(HFC-245fa)气相脱氟化氢制备,由于HFC-245fa是传统发泡剂和热泵工质,氟化工企业无需增加太多的设备投资和工艺开发周期,可直接转产HFO-1234ze(E)。HFO-1234ze(E)的相关研究,更多致力于对气相反应催化剂的改进,高转化率、长寿命的气相脱氟化氢是改进的主要目标。铬基催化剂广泛应用于氟氯交换等反应中,是迄今为止应用最为成功的金属氟化物之一,浙江师范大学罗孟飞教授[44]以铬基催化剂NiO/Cr2O3开展了HFC-245fa脱氟化氢研究,原料转化率接近80%。由于六价铬存在致癌和生物累计性,吕剑团队发明了原位氟化造孔构建大比表面积金属氟化物的方法,分别制备获得了纳米氟化镁催化剂Al/nano-MgF2和nano-AlF3,在此基础上开展了HFC-245fa 脱氟化氢研究,HFC-245fa 脱氟化氢反应在280oC 下运行了200h,非铬基氟化物催化剂显示出了较高的活性和稳定性[44-45]。
为改善HFO-1234ze(E)的绝缘强度,可将HFO-1234ze(E)分子端位氟原子以体积更大的三氟甲基取代,分子为反式-1,1,1,4,4,4-六氟-2-丁烯,其绝缘强度为SF6的1.6 倍,除液化温度偏高外,其余理化与电气性能俱佳[46]。西安交通大学李兴文课题组采用高精度脉冲汤逊装置测量了HFO-1336mzz(E)及其混合气体的电离/吸附速率系数、电子漂移速度等电子输运参数,从微观层面揭示了气体放电机理[47];在不同电场条件下进行了气体击穿实验和局部放电实验,评估了HFO-1336mzz(E)及其混合气体的绝缘性能[48];研究了HFO-1336mzz(E)混合气体作为灭弧介质在12kV 负荷开关中的灭弧特性,采用磁流体动力学模型模拟了压气式负荷开关的电流开断过程,分析了燃弧过程中的电弧电压、灭弧室内温度场、气流场分布以及弧后热击穿特性[49]。以上结果为HFO-1336mzz(E)混合气体作为灭弧介质在中压开关设备中的应用提供了重要参考。
相关HFO-1336mzz(E)制备工艺报道较多,主要制备工艺见图7。杜邦公司[50]以1,1,1-三氯三氟乙烷(R113a)为起始原料,通过异构、气相偶联、加氢脱氯等反应制备得到HFO-1336mzz(E)。该工艺缺点是选用原料为氟利昂,由于破坏臭氧层,R113a已被国际社会列为禁止使用物,因此该工艺受到严格管控。气相异构法也是近年来研究较多的制备HFO-1336mzz(E)的工艺,在酸性催化剂作用下,HFO-1336mzz(Z)能够在高温下异构为HFO-1336mzz(E)[51],但由于原料HFO-1336mzz(Z)价格较高,用于合成HFO-1336mzz(E)显然不经济。Honeywell 公司[52]以四氯化碳和乙烯为原料通过调聚、脱氯化氢、二次调聚和氟化四步反应制备得到了HFO-1336mzz(E)。该工艺选用原料为大宗化学品,反应工艺也均为氟化工通用技术,但由于中间体3,3,3-三氯丙烯稳定性差,容易发生异构、聚合,因此,吕剑团队[53]对此工艺进行了改进,采用3,3,3-三氟丙烯和四氯化碳为原料,通过调聚、气相氟化两步反应制备得到了HFO-1336mzz(E),总收率可达90%以上。

图7 HFO-1336mzz(E)的主要制备工艺
3 SF6替代气体构效关系
近年来,以理论计算驱动的新材料研发模式正在成为新的研究范式。通过分子拓扑几何和电子结构的计算,构建结构与性能之间的关系,快速判断材料的可行性和应用价值,加快新材料的研究步伐。对于环保绝缘气体筛选工作,研究人员最关心的是通过计算得到特定结构分子对应的沸点、绝缘强度、GWP 等性质,判断是否满足替代SF6的需求,有效降低实验合成探索和性能测试的工作量,大大缩短研发周期。其中如何快速准确获取绝缘强度、沸点和GWP 是新型环保绝缘气体分子设计领域的焦点问题。预测方法主要包括物理模型定义、基团贡献、定量构效关系(QSPR)、机器学习等。
3.1 物理模型定义
物理模型定义方法直接从气体的性质定义出发,需要消耗大量计算资源用于量子力学和分子动力学模拟,因此仅适用于对小范围分子的性能预估和筛选。通常情况下高性能计算能力不支持大规模、高通量的分子设计与筛选。
绝缘强度预测方面物理模型定义基于非平衡态等离子体化学理论,考虑气体中电子与重粒子之间的弹性、激发、电离、吸附等碰撞过程,构建以碰撞截面为桥梁的冷态、热态气体临界击穿场强模型,并综合考虑电子动力学和离子动力学的电子崩时空发展模型,得到临界击穿场强与温度、压力、混合比例的定量关系及电击穿协同效应,最终推导出绝缘强度[54]。
沸点预测方面,物理模型定义采用力场描述分子内、分子间的相互作用力,并通过牛顿运动方程计算体系中分子在一定时间尺度下的运动情况,进而模拟出整个体系在一定时间下的热力学、动力学特性。分子动力学方法可以对特定物质的气-液转变过程进行模拟,考察体系密度随温度的变化,确定其转变点(沸点)。该方法被应用于氘、萘、蒽等体系[55]。分子动力学模拟方法预测沸点必须选择精确的力场,但现有力场大多用于描述凝聚态物质的相关性质,力场对气-液转变模拟的泛用性还需根据体系具体分析。同时,液体相变的过程还涉及到相变成核等复杂非平衡过程[56],对特定体系,可能会造成较大误差。
GWP 预测方面, 物理模型定义方程如式(1)[57-58]。
式中,i代表目标物质;TH为GWP的时间范围(通常取值20年、100年或500年);ai为辐射效率;τ为分大气寿命;分母部分AGWPCO2为二氧化碳绝对全球变暖潜能,20年、50年和100年的AGWPCO2分别为0.0243pW·m-1·a·kg-1、0.0895pW·m-1·a·kg-1和0.314pW·m-1·a·kg-1[59]。τ可表示为式(2)。
式中,kOH+CH3CCl3为羟基自由基OH与CH3CCl3的反应速率常数,数值为6.686×10-15cm3/(mol·s)[60];τi为CH3CCl3的大气寿命,数值为5.7 年[61];kOH+i为OH与i物质反应速率常数,可通过Eyring 过渡态理论[62](TST)和Wigner 穿隧效应校正[63]计算。反应速率常数的准确性主要依赖于量子化学计算水平,涉及过渡态的结构优化和频率分析。更精确的反应速率常数需要考虑更复杂的理论,比如变分过渡态理论(VTST)[64]。此外,还需要考虑构象的影响[65]。瞬时辐射效率ai可通过式(3)计算。
式中,Ak为i物质的红外光谱强度;vk为振动频率;F为瞬时辐射强迫。红外光谱可通过量子化学计算获得,对于不同的量子化学计算水平的振动频率需要校正[66]。F(vk)与Ak的关系可参考文献[67-68]。
GWP计算主要依靠根据定义计算的物理模型。基团贡献、QSPR 和机器学习等方法很难描述复杂的反应势能面。根据GWP的定义[式(1)]进行定量计算需要明确化合物的辐射强迫和大气寿命数值,而瞬时辐射强迫可由量子化学方法快速准确地预测,本文主要讨论大气寿命τ或反应速率常数kOH+i的预测。通过量子化学计算反应能垒推导大气寿命或反应速率的方式需要找到反应的过渡态。而过渡态位于势能面上的一阶鞍点,其几何优化的计算量远大于目标化合物。于是发展出一些仅需要量子化学计算目标化合物的方法,如以键解离能替代反应能垒[69-70],以半经验计算获得的分子轨道参数作为反应速率的变量[71-72],从而降低计算量。
3.2 基团贡献
基团贡献法源于研究人员对分子结构与性质最直观的经验,即将特定基团对分子性质的贡献予以量化并加和,借以预测分子性质。在绝缘强度预测方面,2021 年,侯华等[73]以65 种绝缘气体分子为基础,利用DFT 方法获得的表面静电势五参数,关联绝缘强度,建立了构效关系模型,但模型的相关系数仅有0.844。于是侯华等[73]又成立了一种官能团加和计算绝缘强度的方法,理论预测值与实验值的相关系数达到0.9879,优于构效关系模型方法。沸点基团贡献法研究进展见表2。GWP评估的关键在于预测温室气体i与羟基自由基的气相反应速率kOH+i[式(2)],Atkinson等[74-76]基于反应机理及官能团种类、数目等建立了羟基自由基反应速率预测模型。该模型预测值偏差通常不超过一倍,但使用范围有限。Minakata 基于Atkinson 的思路,建立羟基自由基液相反应基团贡献法模型,扩展了该方法的相态适用范围[77]。不难看出,随着基团贡献法的不断改进,在训练集中准确性提升的同时,计算方法越来越繁复,对基团拆分、识别、贡献计算的难度越来越大,容易造成人为误差,无法保证实际应用中的准确性。

表2 基团贡献法预测气体沸点的模型
3.3 QSPR
QSPR 方法是从特定分子结构中提取有关的分子描述符,例如电离能、电子亲和能、分子表面积等,并将其与目标性质建立起函数关系,相比注重表观结构的基团贡献法,QSPR 方法更接近构效关系的物理本质,其优势在于可以通过高通量的量子化学计算获得分子描述符,从而可批量进行大规模的性能预测与筛选。绝缘强度QSPR 研究进展见表3。沸点QSPR研究进展见表4。从以上例子可以看出,不同模型中选用的描述符不同,虽然有些描述符之间在物理意义上相同,但是缺乏统一描述气体沸点构效关系的模型及描述符。各种预测模型有些相关系数较低,有些相关系数虽然较高,但缺乏测试集验证,在绝缘强度预测时尤为突出。QSPR方法研究结果再次证明物质的沸点受其分子的大小(基团贡献法中体现为分子量、基团数、原子数等,QSPR 法中体现为分子表面积等)、分子极性(基团贡献法中体现为极性基团个数,QSPR 法中体现为分子极化率、电离能、表面电荷分布情况、化学硬度等)等因素影响,分子越大、极性越高,其沸点越高;同时,提高气体的绝缘性能往往也需要分子具有更大表面积、更强的极性(意味着更大的正电势表面积比例和更强的正电势数值)。沸点和绝缘强度对分子结构的要求形成了矛盾,所以在绝缘气体分子设计中,必须考虑平衡两个指标。此外,QSPR 方法还可用于预测kOH+i[式(2)],以获得绝缘气体的GWP。美国国家环境保护局开发的软件ESI suite中,其AOPWIN模块基于原子中心片段方法,结合内置反应速率数据集,对原子中心片段最相似的五个参考物质进行加权计算[78],实现了反应速率常数kOH+i的QSPR 方法预测[79]。这种方法计算过程较为复杂,且准确性取决于参数数据的完备性。通过提取目标物质i的量子化学、Dragon 等描述符,基于最小二乘拟合、多元线性回归、随机森林算法、提升决策树等方式建立kOH+i与描述符的关系,是近年来QSPR 预测kOH+i的基本思路。Wang等[80]建立的模型采用22 个描述符,预测均方根误差为0.430,但仅适用于室温。Li 等[81]搜集了不同温度下1543组kOH数据,构建了包含温度参数的关系方程式,扩展了QSPR模型的温度适用范围。

表3 QSPR预测绝缘强度的模型

表4 QSPR预测气体沸点的模型
3.4 机器学习
随着人工神经网络(ANN)等机器学习技术的发展,其在绝缘强度和沸点预测方法上也受到了研究人员的重视。在绝缘强度预测方面,2021 年,邱馨仪[95]以47 种气体绝缘强度为数据集,对比了多元线性回归和误差反向传播ANN 两种方法在建立构效关系模型的能力,发现相关系数R分别为0.954 和0.995 的ANN 拟合效果更佳。在沸点预测方面,2020年Sun等[96]利用ANN 和随机森林(RF)算法分别以66 种气体沸点为数据集构建模型,结果表明ANN 和RF 的模型的拟合度分别为R2等于0.951和0.966。可以看出,对于特定分子集合,经机器学习训练出的模型已经可以达到较好精度,但以上两个工作模型数据集较小,其泛用性尚需提高。2022 年,Liu 等[97]利用决策树(EDT)方法结合ANN对334种制冷剂分子的沸点构建了模型,训练集中的分子包括不饱和键以及—F、—Cl、—Br、—I 等16 种基团,模型拟合度R2=0.951。该工作中的制冷剂分子训练集与绝缘气体有一定的交叉,故而对绝缘气体设计有一定指导作用。机器学习的应用使得研究人员能够建立拟合度较高的模型,但所构建的模型往往包括数量众多的描述符,难以建立描述符与分子性质的物理联系。同时,机器学习需要大量的数据支撑,使模型不断优化,相比之下绝缘气体的训练集规模较小,拟合出模型的泛用性受限。
4 SF6替代气体突破瓶颈的新思路及未来发展趋势
4.1 高通量分子设计
现有分子设计工作筛选空间基本不超过10 万个分子,且难以平衡计算效率、精确性和泛用性,因此,为了挖掘符合目标要求的新型绝缘气体,需要开展高通量分子设计与性能预估。一方面,海量筛选已知化合物,获得分子合成难度小的结构。提取ZNIC2[98]和GDB-17[99]等数据库中的结构编码,进行元素和子结构筛选缩小目标结构范围,然后通过性能预估锁定目标结构[100-101]。另一方面,通过骨架加基团的方式构造未知化合物,在制定合理的化学配位体系下,对小分子采用穷尽所有空间取向的方式构造新型分子结构[102-105]。该方式生成的未知结构数量远多于已知结构,进一步加大了分子结构的搜索空间。发展快速、准确和适用范围广的性能计算方法,以适应高通量分子筛选需求。通过机器学习实现逆向设计,对结构进行打分取长补短,实现性能到结构的自主学习,从而降低全空间搜索的机时代价。为适应高通量计算的需求,开发自动化程序实现从批量输入二维结构到输出性能数据,结合深度学习处理计算过程中的报错和异常结构,并调整参数获得理想的计算结果。
采用如图8所示的高通量分子设计流程,初步设计出3种性能优异的亚胺化合物(表5)。将C≡N 键改为C= = N 键后,不仅能够保持绝缘强度,毒性也随之降低。通过高通量分子模型,设计出的亚胺类分子,可能是未来的研究对象。

表5 具有应用潜力的亚胺化合物

图8 高通量分子设计筛选策略
氟代亚胺(R1R2C= = NF)的制备方法,以腈类与氟化氯、氟气为原料,经加成、脱氯两步反应进行[106]。由于混合气与乙腈等反应则过于剧烈,因此这种方法只适用于卤代腈类。随着全氟烷基链的增加,反应时间也需要相应延长,反应收率可达90%以上。氟化氯的作用不仅能够提高反应活性,由于N-氯代三氟甲胺(CF3NFCl)脱氟化氯难以实现(N—Cl键往往会降解为N—H键),氯原子的引入对第二步脱氯更为重要,研究显示,在三氟乙酸酐中脱氯,产物纯度和收率都有明显改善。
为改善N-氟代二氟甲基亚胺(F2C= = NF)的绝缘强度,可用体积更大的卤素原子或三氟甲基取代氟原子。N-氯代二氟甲基亚胺也可以通过类似工艺制备;将F2C= = NF 与F2C= = NBr 混合后,在碱金属氟化物中发生二聚反应,产物经光照可获得N-三氟甲基二氟甲基亚胺(F2C=NCF3)[107-109],与N—F键的高活性相比,F2C=NCF3特有的内亚胺结构化学稳定性更好。
将N-氟代二氟甲基亚胺(F2C= = NF)链长增加,绝缘强度也可提高至1.17,全氟二亚胺(FN= =CF—CF= = NF)的制备与单亚胺类似[110],原料采用乙二腈与氟化氯、氟气混合,经加成、脱氯即可制备。
4.2 共沸绝缘气体的新方案
由于难以找到兼顾液化温度和绝缘性能的环保气体,使用绝缘强度优异但沸点较高的电负性气体与低沸点缓冲气体组成的混合物,以获得与SF6相似的绝缘强度和沸点,是目前环保气体的解决方案[111]。现有的混合气体为理想气体的“线性加和”,但电负性气体的挥发性远低于稀释气体,更易于液化[17],当混合气体受到扰动(如气体泄漏),电负性气体液化会导致气相密度迅速下降,从而降低气体的击穿电压,引发设备故障。因此,绝缘性能的强协同作用增强了击穿电压对液化的敏感性,给电气设备的安全运行带来了额外的风险[112]。稀释气体的绝缘强度低、与绝缘气体的分子间作用力弱、无法有效调和气体之间液化和绝缘性质的固有矛盾,是现有混合气体的本质缺陷。
正共沸混合物的液化温度低于每种组元的沸点,且共沸物的宏观性质类似单一气体,可有效克服因系统泄漏而导致的组分变化问题[113]。因此,研发共沸绝缘气体,利用多元分子的各自优势,弥补单一分子缺陷[114-115],有望突破技术瓶颈,开发出综合性能优异的SF6替代气体(图9)。
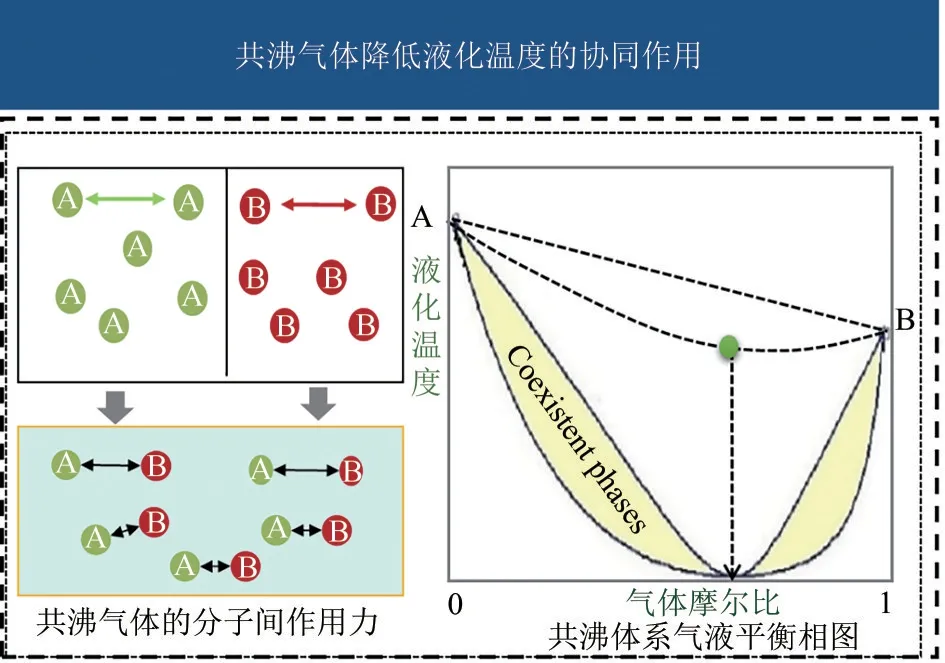
图9 共沸气体的协同作用
共沸物的研究主要包括共沸形成机理和汽液相平衡行为两个方面。虽然现阶段共沸作用的形成机理和调控机制尚缺乏完善的理论[116-118]。但现有的研究表明,同一物质分子间的自缔合作用或不同物质分子间的相互作用(如氢键、二氢键、强范德华力、π-π 作用等)可能是产生共沸现象的主要原因[119]。
国内外学者在汽液相平衡实验测量和理论预测方法方面取得丰富的研究成果。实验测量和理论预测研究汽液相平衡的两种主要途径。实验测量是获得汽液相平衡最直接的途径,法国Centre of Thermodynamics of Processes(CTP)研究中心、中国科学院物理化学研究所、西安交通大学均等国内外机构研制了高精度汽液相平衡测量装置,获得了大量的共沸体系相平衡数据[120-121]。但实验测量的时间、人力、资金成本巨大,不适于共沸体系的筛选。得益于先进算法和计算机技术的发展,基团贡献模型和分子模拟已逐渐成为汽液平衡预测的可靠途径。基团贡献模型认为溶液中的分子是由多个基团组成,分子间的相互作用源自于基团之间的相互作用。若已知分子的基团参数以及基团间的相互作用参数,可以在没有实验数据的条件下,对混合物的汽液平衡进行准确的预测。现有的模型中,最具代表性是UNIFAC[122-123]和PPR78[124]基团贡献模型。分子模拟是预测汽液相平衡的另一重要手段,分子动力学和蒙特卡洛(Monte Carlo)模拟是汽液相平衡分子模拟的主要方法[118,125]。
综上,虽然共沸作用的形成机理还有待从分子相互作用的角度进行解释,但汽液相平衡的研究已取得了非常大的进步,现有的基团贡献模型和分子模拟为共沸体系的开发提供了可靠、有限的途径。建立共沸体系能够使混合气体在降低液化温度和提高绝缘性能两个方面同时产生协同作用,从而弥补现有气体“线性加和”的缺陷,可能成为未来环保绝缘气体的重要研究方向。
5 结语
本文针对目前电力行业SF6替代的迫切问题,以绝缘气体的筛选要求为出发点,回顾了替代气体的探索历程和构效关系模型发展趋势,探讨了现有筛选方法的局限性。从分子结构的角度分析了绝缘强度、液化温度、温室效应等性能的相互影响,从应用角度剖析了研制绝缘气体的难点和关键问题,指出揭示气体的物理化学本质、突破新型绝缘气体的构建策略是环保绝缘气体研发的核心问题,并提出如下解决途径。
(1)将高通量分子设计、材料基因工程等前沿技术引入到新型气体的探索研究中,实现由“经验指导实验”传统模式向“理论预测、实验验证”高效新模式的转变,是探索新型环保气体的有效手段。
(2)将共沸理论应用于绝缘气体的研发,利用共沸气体的分子间作用力降低液化温度、提高绝缘强度、维持组分恒定,有望突破单一气体宏观性质的固有矛盾,开发出综合性能优异的环保混合气体。
(3)通过绿色催化反应工艺和高效低耗的化工过程协同创新,助力实现新型绝缘气体从气体设计、气体制造到气体应用的全生命周期绿色变革,有望引领气体绝缘技术的源头创新。
猜你喜欢
数学物理学报(2021年5期)2021-11-19
数学物理学报(2020年4期)2020-09-07
小哥白尼(趣味科学)(2018年11期)2018-12-18
幸福(2018年33期)2018-12-05
铁道通信信号(2018年6期)2018-08-29
铜仁学院学报(2018年6期)2018-07-05
现代传输(2016年4期)2016-12-01
衡阳师范学院学报(2016年3期)2016-07-10
焊接(2015年8期)2015-07-18
浙江理工大学学报(自然科学版)(2015年5期)2015-03-01
