
CDK5对胃癌PD-L1表达及生物学特性的影响
2023-09-25 00:50:44黄谊强王子明罗杨罗水妹谢贤和张帆
中国医科大学学报 2023年9期
黄谊强,王子明,罗杨,罗水妹,谢贤和,2,3,张帆,2,3
(1.福建医科大学附属第一医院肿瘤内科,福建医科大学分子肿瘤研究所,福州 350005;2.福建医科大学附属第一医院滨海院区国家区域医疗中心肿瘤内科,福州 350212;3.福建省肿瘤精准诊疗重点实验室,福州 350005)
胃癌恶性度高,全球发病率和死亡率分别高居癌症的第5位和第4位[1]。近年来,PD-1/PD-L1抑制剂对恶性肿瘤 (如恶性黑色素瘤、肺癌等) 的治疗效果取得了里程碑式的进展[2],但在胃癌方面却仍未获得预期疗效[3]。因此,探讨调控胃癌PD-L1表达进而抑制其介导的免疫逃逸的新策略至关重要。CDK5既往被认为主要在神经系统中发挥作用,近年来发现CDK5参与肿瘤的免疫、凋亡、增殖、转移等[4-7],是肿瘤靶向治疗及免疫治疗的潜在靶点。研究[4,8]表明,CDK5可调控PD-L1,在不同类型的肿瘤中机制也不尽相同,而其在胃癌中的调控作用尚未明确。本研究拟探讨CDK5对胃癌PD-L1表达及肿瘤增殖、迁移等特性的影响,以及抑制CDK5在胃癌免疫治疗及靶向治疗中的可行性。
1 材料与方法
1.1 数据来源
通过癌症基因组图谱 (The Cancer Genome Atlas,TCGA) 数据库 (https://portal.gdc.cancer.gov/) 下载375例胃癌样本和32例正常组织样本的转录组每千个碱基的转录每百万映射读取的片段数 (fragments per kilobase of exon model per million mapped fragments,FPKM) 数据及相应的临床病理和生存资料。
1.2 实验材料及方法
1.2.1 主要试剂和仪器:人胃癌细胞株BGC-823购自上海翼和应用生物技术有限公司;CDK5抑制剂20-223购自美国MedChemExpress公司;CDK5过表达质粒委托北京擎科生物公司合成;PD-L1抗体购自美国Proteintech公司;β-actin抗体购自英国Abcam公司。凝胶成像仪 (Biosciences AccuriC6型) 购自美国Bio-Rad公司。
1.2.2 细胞培养:用含15%胎牛血清、1%双抗 (青霉素1 000 U/mL、链霉素0.1 mg/mL) 的DMEM培养基,于37 ℃、5%CO2恒温培养人胃癌BGC-823细胞。
1.2.3 CCK-8法:测定CDK5 抑制剂 20-223 分别作用24、48、72、96 h对细胞活力的影响及其IC50。
1.2.4 Western blotting:20 μmol/L CDK5抑制剂处理胃癌BGC-823细胞24 h,按常规方法提取细胞总蛋白。上样 (每泳道20 μg) 后,行聚丙烯酰胺凝胶电泳。转至 0.45 μm PVDF膜,室温封闭1 h。4℃孵育一抗过夜,TBST洗膜,室温孵育二抗1 h,TBST洗脱,ECL化学发光显影。β-actin作为内参照。导出照片后用Adobe Photoshop软件读取各条带灰度值。
1.2.5PD-L1表达检测:1/1 000DMSO及0、0.16、0.31、0.63、1.25、2.50、5.00、10.00、20.00、30.00、40.00 μmol/L CDK5抑制剂作用胃癌BGC-823细胞24 h,实时定量聚合酶链式反应 (real-time fluorescence quantitative polymerase chain reaction,RT-qPCR) 检测PD-L1表达;10 ng/mL干扰素-γ (inteferon-γ,IFN-γ) 5 μmol/L CDK5抑制剂、10 ng/mL IFN-γ+5 μmol/L CDK5抑制剂分别作用于胃癌BGC-823细胞24 h,qPCR检测PD-L1表达;用CDK5过表达质粒、psPAX2质粒及VSVG质粒构建CDK5过表达稳转株,qPCR检测PD-L1表达。
1.2.6 细胞周期检测:20 μmol/L CDK5抑制剂分别抑制胃癌BGC-823细胞24、48、72、96 h,碘化丙啶染色,流式细胞术检测细胞周期。
1.2.7 细胞凋亡检测:80、40 μmol/L CDK5抑制剂抑制胃癌BGC-823细胞CDK5,Annexin V-FITC/碘化丙啶染色,流式细胞术检测细胞凋亡。
1.2.8 细胞迁移趋化能力检测:2 μmol/L CDK5抑制剂作用胃癌BGC-823细胞24 h,Transwell 测定细胞迁移趋化能力;1 μmol/L CDK5抑制剂作用胃癌BGC-823细胞48 h,划痕实验测定细胞迁移能力。
1.2.9 细胞增殖检测:0.02、0.04 μmol/L CDK5抑制剂作用胃癌BGC-823细胞2周,克隆形成实验检测细胞增殖能力。
1.3 统计学分析
采用GraphPad Prism 8进行统计分析,计量资料2组间比较采用t检验,2组以上样本数据采用单因素方差分析及两两比较。P< 0.05为差异有统计学意义。
2 结果
2.1 CDK5及PD-L1的表达
对TCGA数据库的中CDK5表达量进行泛癌分析,结果显示,CDK5在多种癌症中均呈高表达,见图1A。对胃癌CDK5及PD-L1表达量进行分析,发现配对样本及非配对样本CDK5及PD-L1表达量均显著高于正常样本,见图1B~1E。

图1 CDK5与PD-L1差异表达分析Fig.1 Differential expression analysis of CDK5 and PD-L1
2.2 CDK5与胃癌分期、免疫检查点、免疫细胞相关
构建CDK5高、低表达组差异基因热图及临床特征热图,对CDK5与免疫检查点 (包括PD-L1)、免疫细胞浸润进行相关性分析,结果显示,CDK5与胃癌免疫检查点、免疫细胞浸润和N分期相关。
2.3 CDK5高、低表达组的基因本体 (Gene Ontology,GO) 功能分析和京都基因和基因组数据库 (Kyoto Encyclopedia of Genes and Genomes,KEGG) 通路富集分析
GO分析表明,CDK5显著富集的生物过程包括肌肉收缩、肌肉细胞分化等。显著富集的细胞成分包括神经元胞体、突触前结节肌节、肌肉纤维等。显著富集的分子功能包括离子通道活性、被动膜转运体活性肌肉结构成分等。
KEGG通路富集分析发现,差异基因富集通路有钙离子信号转导通路、cAMP信号转导通路、cGMP-PKG信号转导通路、胃酸分泌通路等。
2.4 CDK5抑制剂对胃癌BGC-823细胞活力的影响
检测不同浓度CDK5抑制剂20-223对BGC-823细胞活力的影响,结果显示,24、48、72、96 h的IC50分别为131.70、40.72、1.33、0.90 μmol/L,见图2。
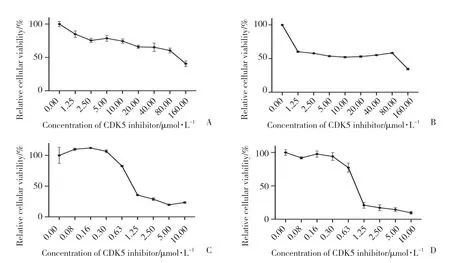
图2 CDK5抑制剂20-223对胃癌BGC-823细胞的剂量-反应曲线Fig.2 Dose-response curve of CDK5 inhibitor 20-223 on gastric cancer BGC-823 cells
2.5 抑制及过表达CDK5对PD-L1表达的影响
应用CDK5抑制剂对胃癌BGC-823细胞干预24 h,Western blotting及qPCR检测显示,PD-L1蛋白及mRNA表达显著下调 (P= 0.013 4,P= 0.000 4)。应用不同浓度CDK5 抑制剂 20-223对胃癌BGC-823细胞干预24 h,发现2.5 μmol/L抑制剂可最大程度抑制PD-L1转录。见图3A~3C。
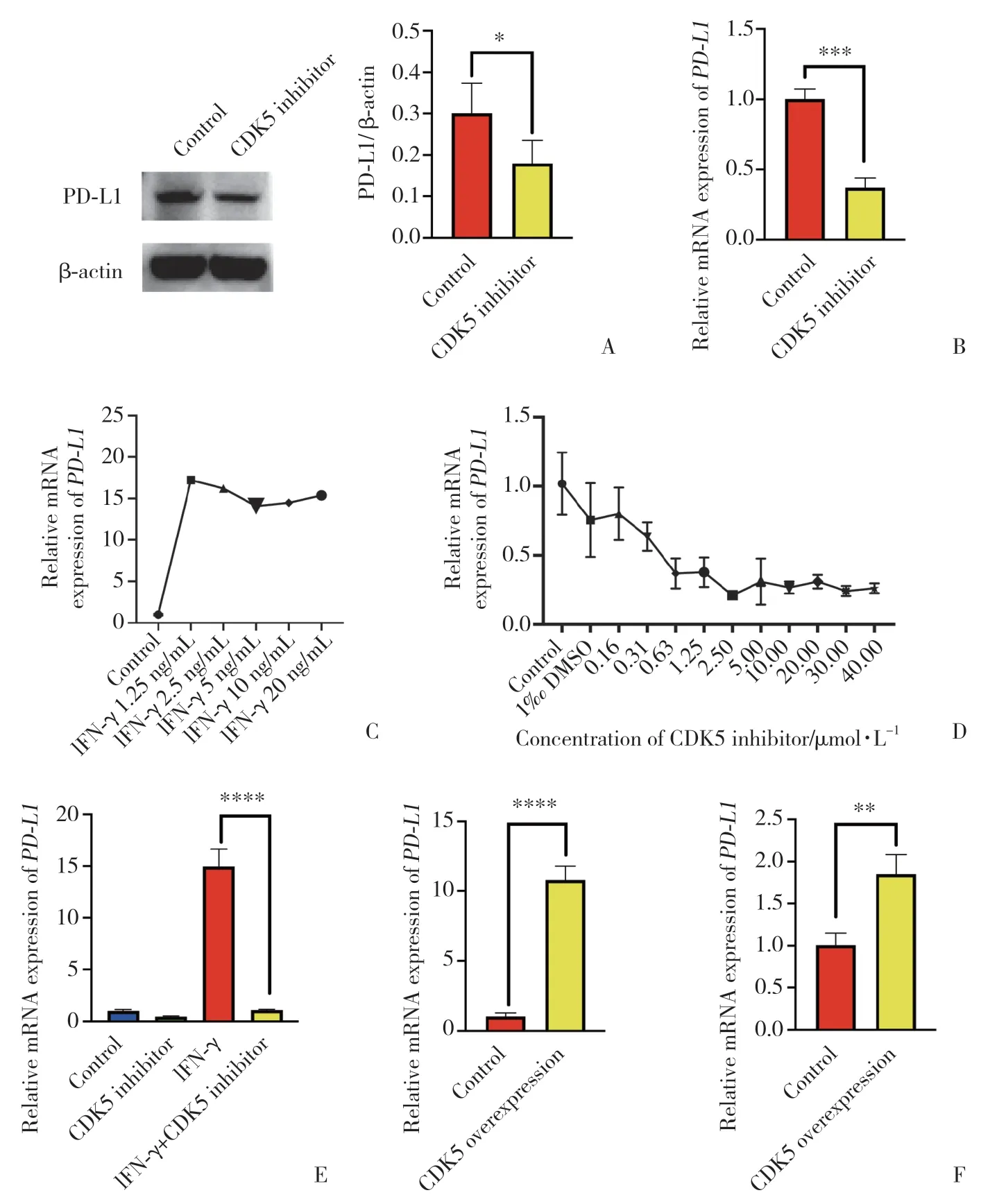
图3 CDK5调控PD-L1表达及其机制.3 CDK5 regulates PD-L1 expression and its mechanism
2.6 CDK5抑制剂挽救IFN-γ促进的PD-L1转录
IFN-γ可显著促进PD-L1转录,1.25 ng/mL IFN-γ可最大程度促进PD-L1转录,而CDK5抑制剂可极大程度挽救IFN-γ促进的PD-L1转录,提示CDK5通过IFN-γ激活的通路下调PD-L1表达。见图3D。
2.7 过表达CDK5促进PD-L1转录
CDK5过表达效率达10.80倍;PD-L1相对表达值升高1.85倍。见图3F。
2.8 CDK5抑制剂对胃癌BGC-823细胞周期的阻滞作用
应用20 μmol/L CDK5抑制剂干预胃癌BGC-823细胞,流式细胞术周期检测结果显示,与对照组相比,CDK5抑制剂组细胞周期被抑制在G2/M期,干预12 h、24 h、48 h时,G2/M期细胞比例呈递增趋势,见图4。

图4 抑制CDK5致胃癌BGC-823细胞周期抑制于G2/M期Fig.4 Inhibition of CDK5 in gastric cancer BGC-823 cell cycle arrest in G2/M phase
2.9 抑制CDK5促进胃癌BGC-823细胞凋亡
应用80、40 μmo/L CDK5抑制剂干预胃癌BGC-823细胞48 h,流式细胞术周期检测结果显示,与对照组相比,40 μmol/L组早期凋亡及晚期凋亡均无明显差异,但总凋亡细胞比例增加 (P= 0.037 7),80 μmol/L组较40 μmol/L组进一步增加 (P= 0.000 5);80 μmol/L组早期及晚期凋亡细胞比例均增加 (P=0.000 2,P= 0.003 9)。以上均提示抑制CDK5可促进细胞凋亡。见图5。
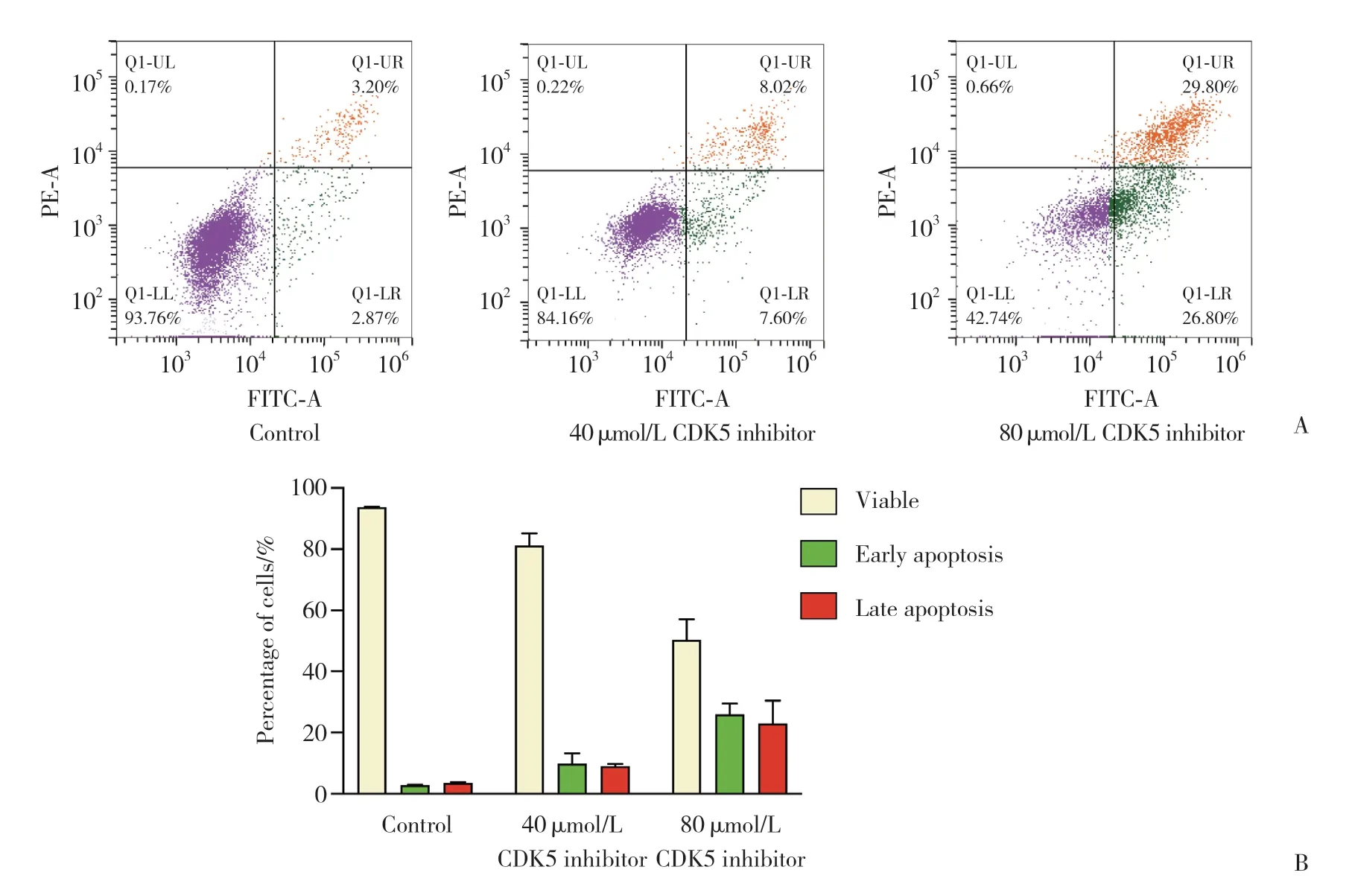
图5 抑制CDK5对胃癌BGC-823细胞凋亡的影响Fig.5 Effects of inhibiting CDK5 on the apoptosis of gastric cancer BGC-823 cells
2.10 抑制CDK5减弱胃癌BGC-823细胞迁移能力
划痕实验结果显示,CDK5抑制剂组迁移率明显低于对照组 (4.39% vs 24.37%,P= 0.002 6),表明抑制CDK5使胃癌BGC-823细胞迁移趋化能力明显减弱。见图6A、6B。

图6 抑制CDK5对胃癌BGC-823细胞迁移能力的影响Fig.6 Effects of inhibiting CDK5 on the migration ability of gastric cancer BGC-823 cells
2.11 抑制CDK5降低胃癌BGC-823细胞迁移趋化能力
Transwell实验结果显示,CDK5抑制剂组细胞穿过数量明显低于对照组 (20倍物镜下15.20个/视野 vs 65.87个/视野,P= 0.002 5),表明抑制CDK5可使胃癌BGC-823细胞迁移趋化能力明显减弱。见图6C、6D。
2.12 抑制CDK5降低胃癌BGC-823细胞增殖能力
与对照组 (克隆形成率80.87%) 相比,CDK5抑制剂0.02 μmol/L组克隆形成能力 (80.13%) 无明显改变 (P= 0.996 1),0.04 μmol/L组克隆形成能力(32.60%) 则明显降低 (P= 0.000 4)。见图7。

图7 抑制CDK5对胃癌BGC-823细胞克隆形成能力的影响Fig.7 Effects of inhibiting CDK5 on the colony formation ability of gastric cancer BGC-823 cells
3 讨论
肿瘤主要表现为肿瘤细胞增殖增加,侵袭增强,凋亡减少,代谢特征和免疫微环境改变,血管结构异常,以及免疫逃逸等[9]。对应这些肿瘤特性,本研究探索了CDK5对胃癌BGC-823细胞免疫检查点PD-L1及细胞周期、凋亡、迁移的影响。
本研究结果显示,抑制胃癌BGC-823细胞CDK5表达可致PD-L1mRNA和蛋白表达均下调,过表达CDK5则可促进PD-L1mRNA表达,与本研究中生物信息学预测提示的“CDK5及PD-L1在胃癌中均高表达,且二者呈正相关”一致,符合转录调控或转录后调控的特点。
胃癌中,PD-L1 表达主要受IFN-γ/JAK/STAT1/IRF1通路转录调控[10]。IRF1主要由磷酸化STAT1转录促进调控,而JAK、STAT的活性都通过磷酸化调节,是CDK5作用的潜在位点。本研究发现,抑制胃癌BGC-823细胞CDK5可挽救IFN-γ刺激的PD-L1mRNA表达,提示CDK5在胃癌BGC-823细胞中通过IFN-γ通路转录促进PD-L1表达。在髓母细胞瘤中,CDK5也通过IFN-γ通路调控PD-L1,CDK5并非直接作用于此通路引起JAK、STAT1磷酸化水平及IRF1表达水平改变,而是通过IRF2BP2/IRF2/IRF1途径转录促进PD-L1表达,但CDK5直接作用的位点尚未明确[4]。PTPN2、SOCs、PTP1B、PIAS等多种分子均可作用于JAK/STAT 通路,调控IFN-γ诱导的基因 (如PD-L1) 表达,可能是胃癌CDK5间接作用于IFN-γ/JAK/STAT通路促进PD-L1表达的潜在旁路[11-14]。
在肺腺癌中,敲低CDK5可下调PD-L1表达,但PD-L1mRNA水平却没有改变,表明CDK5不是在转录水平调控PD-L1,而是通过抑制TRIM21泛素化活性,抑制PD-L1泛素化降解,最终导致PD-L1上调[8]。故胃癌BGC-823细胞中CDK5调控PD-L1机制与肺腺癌中不同。表明不同癌症中CDK5调控PD-L1的方式不尽相同。
转录因子是转录调控的关键,PD-L1启动子报告基因可检测转录水平的改变,CDK5的直接作用分子及其磷酸化位点则需要蛋白质谱磷酸化作用分析、蛋白质谱相互作用分析并结合测序通路分析进一步探索,并最终进行体外验证。
综上所述,本研究结果显示,CDK5与PD-L1在胃癌中均高表达且呈正相关,在胃癌BGC-823细胞中,CDK5通过IFN-γ通路促进PD-L1表达,CDK5抑制剂在下调PD-L1的同时,促进细胞凋亡、阻滞细胞周期、降低细胞迁移能力及克隆形成能力。
猜你喜欢
国际呼吸杂志(2019年4期)2019-03-12 01:07:30
中国报道(2018年2期)2018-04-20 04:12:46
上海农业学报(2017年3期)2017-04-10 12:39:26
金融博览(2016年7期)2016-08-16 18:44:41
绿色中国(2016年1期)2016-06-05 09:02:59
中华老年多器官疾病杂志(2016年7期)2016-04-28 08:43:05
癌症进展(2016年10期)2016-03-20 13:15:43
医学研究杂志(2015年5期)2015-06-10 06:43:26
中国当代医药(2015年16期)2015-03-01 02:03:13
软件工程(2014年6期)2014-09-24 12:23:56
