
基于BSA-seq方法挖掘大豆再生相关候选基因
2023-09-14 09:15赵宇晶张滨烁苏安玉于振海李佳欢张艳婷武小霞
作物学报 2023年11期
赵宇晶 张滨烁 苏安玉 于振海 李佳欢 林 洋 张艳婷 武小霞,* 赵 莹,*
基于BSA-seq方法挖掘大豆再生相关候选基因
赵宇晶1,**张滨烁1,**苏安玉2于振海1李佳欢1林 洋1张艳婷1武小霞1,*赵 莹1,*
1东北农业大学农学院, 黑龙江哈尔滨 150030;2东北农业大学资环学院, 黑龙江哈尔滨 150030
转基因生物育种技术可以定向改良大豆品种, 为大豆定向育种提供一种新思路。为寻找与大豆再生相关的基因, 探索大豆再生规律、提高遗传转化效率, 本研究利用再生能力强材料东农50、再生能力弱材料Keburi及其子代RIL群体的200份材料进行大豆器官发生试验, 比较不同基因型之间再生能力的差异, 筛选出极端材料各20份, 后通过BSA-seq (bulked segregant analysis sequencing)技术对大豆再生候选基因进行初步定位, 共获得88.04 G的clean data, 平均测序深度为20.03×, 定位到2 Mb区间, 利用GO等数据库对区间内基因进行富集分析, 差异表达基因主要富集在纤维素微纤维组织、植物型细胞壁组织或生物发生等20个条目中, 其中被显著富集的植物型细胞壁组织或生物发生条目中共有6个基因, 对6个基因进行组织表达量分析, 在丛生芽伸长期间表达水平较高, 说明其在大豆再生过程中发挥作用, 可能为影响大豆再生的关键基因。本研究为大豆再生机制研究提供了重要的候选基因信息与必要的材料基础。
大豆; 再生; 器官发生; BSA-seq
大豆((L.) Merr.)是世界最大的植物蛋白来源和重要的食用油脂来源。能够为人类提供优质的植物蛋白。大豆作为世界上种植面积最大的转基因作物, 转基因大豆的种植面积逐年增加, 占到全部转基因作物的50%以上[1]。但目前, 大豆遗传转化效率较低, 主要是由于大豆的再生系统不稳定, 其中再生效率和频率受基因型影响明显[2-4], 这就限制了将遗传转化生物技术应用在大豆遗传改良上。因此, 寻找和建立易操作、高效率、稳定性好的大豆再生体系是提高大豆遗传转化效率的重要前提和保障[5]。器官发生途径是指通过外植体诱导产生丛生芽后丛生芽伸长形成根, 最终形成完整植株的过程。该途径具有诸多优点如外植体容易获得, 不受季节限制等。Cheng等[1]在高浓度6-BA的培养基中放置大豆子叶节后诱导出丛生芽, 获得再生植株; Kartha[6]采用栽培大豆真叶通过器官发生来再生植株; Paz等[7]使用大豆的半种子作为外植体, 使大豆子叶节技术进一步改进, 简化了流程, 提高了大豆的转化效率; 王怡婷等[8]采用过氧化氢对种子进行消毒改进种子灭菌方法; Li等[9]对农杆菌介导的大豆转化提供了改进的方案, 优化了激素浓度组合。
近年来, 植物的再生机制研究受到人们的广泛关注, 对再生基因的研究报道也越来越丰富, 在此前的研究中已经发现并克隆了一些影响植物再生的基因, 植物干细胞拥有自我更新的能力并不断分化, 主要位于根尖分生组织(root apical meristem, RAM)和茎尖分生组织(shoot apical meristem, SAM), 而植物在形态过程中的细胞都是来源于植物干细胞, 干细胞对于调控植物的再生也就具有重要意义。植物再生离不开干细胞对与SIM和SEM的调控,基因、和基因作为植物干细胞的明星基因, 也就被当作提高植物再生能力的关键。最早发现的基因作为干细胞的最有明显功能的基因[10], 将基因作为标记后可以将植物内的细胞分为干细胞和非干细胞。1996年首次报道的STM蛋白是分生组织内干细胞的激活和关键调控因子[11-12]。2002年通过遗传学手段发现基因在胚胎发育过程中起着关键作用[13], 发现在拟南芥上是干细胞调控的正调控的转录因子[14]。Mayer等[15]在拟南芥中分离得到了基因, 发现了参与植物分生组织的形成。而3个核心基因是如何调控植物干细胞的也就成为了大家关注的核心, 近几年来大家也陆续发现三者有着精密的互作模式, 对于植物分生组织的干细胞调控有着重要意义。2020年发现可以调控和激活的转录, 并与相互作用,也可以激活的表达,通过和的相互作用可以负调控基因的转录, 从而可以控制干细胞的大小[16]。评定一个植物再生能力的好坏不定芽的数目也是关键, 在拟南芥中发现作为ERF家族的转录因子,可以与它的同源基因可以促进芽的再生, 证明了可以促进茎尖分生组织的形成[17]。Banno等[18]研究表明过量表达基因可以增加不定芽的数目。而植物的再生同样也离不开体细胞胚的发育,作为体细胞胚的关键调控的转录因子。蔡英卿[19]发现可以促进体细胞胚胎发育, 将进行拟南芥转化后发现可以使拟南芥再生促进植物的恢复[20],可以促进细胞的全能性, 有研究表明可以调控2个细胞全能性的关键调节因子LEC1和LEC2, 同样也发现介导的体细胞胚需要基因的帮助[21]。并且, 李思楠等[22]通过体细胞胚试验筛选到与再生相关的基因并证实其促进再生。
近几年来随着测序手段的提高与成本的下降, 使得全基因组测序的BSA (bulked segregant analysis)方法可以用来进行挖掘候选基因, 被广泛应用于大豆[23-27]、番茄[28]、水稻[29-30]、黄瓜[31]等作物基因定位与基因挖掘。然而, 到目前为止BSA-seq技术应用于大豆再生相关基因的挖掘研究尚未见报道。
本研究采用器官发生途径筛选200份群体材料, 对父母本及后代2个极端混池共4份材料使用BSA技术, 通过全基因组测序, 定位与再生性状相关联的区域, 对区域内候选基因进行生物信息学分析、表达模式分析, 旨在筛选再生能力强的基因型及与大豆再生相关的基因, 提高大豆遗传转化效率。
1 材料与方法
1.1 试验材料
本研究所使用的RIL群体材料于2017年以东农50 (东北农业大学, 哈尔滨)为母本, 日本品种Keburi (日本)为父本杂交后进行自交, 逐年筛选农艺性状稳定的材料进行种植, 得到稳定200份的重组自交系(recombinant inbred lines, RILs)材料。本试验使用F5代自交种子材料进行器官发生试验。
1.2 大豆器官发生试验
挑选表面光滑、无病斑、大小一致的大豆种子放入灭菌后的锥形瓶中, 使用75%的无水乙醇浸泡5 min对种子表面进行消毒, 后倒入15%过氧化氢放入37℃摇床中110转 min–115 min。用无菌水冲洗5~6次至表面干净无浮沫。倒入约200 mL无菌水浸泡, 常温避光放置24 h。
将浸泡好的种子沿下胚轴切开, 一分为二, 下胚轴切至2~3 mm, 切除腋芽, 将切好的外植体下胚轴朝下插入SIM丛生芽诱导培养基中培养14 d, 然后将长出丛生芽移至SEM丛生芽伸长培养基中, 放置14 d后继代一次。将伸长的苗放置在RM生根培养基中等待生根, 统计数据。大豆器官发生试验过程如图1所示。
培养基配方如下。SIM丛生芽诱导培养基: 3.21 g mL–1B5+0.6 g mL–1MES+30 g mL–1蔗糖, pH 5.7~5.8灭菌后加入B5、6-BA; SEM丛生芽伸长培养基: 4.33 g mL–1MS+0.6 g mL–1MES+30 g mL–1蔗糖, pH 5.7~5.8灭菌后加入B5、IAA、GA3、Glu、Asp; RM生根培养基: 1.605 g mL–1B5+0.6 g mL–1MES+20 g mL–1蔗糖, pH 5.8灭菌后加入B5、Asp、Glu。

图1 器官发生试验流程图
A: 萌发; B、C: 诱导丛生芽; D: 丛生芽伸长; E、F: 生根培养。
A: seed germination; B, C: clustered bud induction; D: clumps of buds elongated; E, F: rooting culture.
1.3 表型分析
根据材料的生长情况, 统计各项数据, 对再生情况进行鉴定, 各试验材料每个处理20个外植体, 2次重复, 以下为统计指标。再生潜力(regenerative potential, RP): 丛生芽数/外植体数; 再生能力(regenerative ability, RA): 伸长苗数/丛生芽数; 再生效率(regenerative efficiency, RE): 伸长苗数/外植体数; 再生率(regenerative rate, RR): 生根数/外植体数。
采用SPSS 21.0将4个指标综合计算特征值、贡献率、主成分、隶属函数及值。根据值筛选强再生和弱再生的材料各20 份。
1.4 BSA混池定位
选取双亲、20份强再生材料和20份弱再生材料的幼嫩叶片, 提取DNA, 样品基因组进行琼脂糖凝胶电泳检测, 将检测合格后的DNA进行1∶1的混合, 利用超声波将亲本和混池DNA序列形成随机片段, 对随机片段化的DNA依次进行末端修复、3¢端加A、连接测序接头后, 再利用磁珠吸附富集基因组长度350 bp左右的片段, 经过PCR扩增形成测序文库。后根据欧式距离(Euclidean Distance, ED)算法评估与性状关联区域[32]。ED值越高, 代表该点关联效果越好。
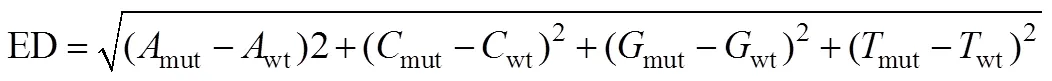
式中,mut为A碱基在突变混池中的频率,wt为A碱基在野生型混池中的频率;mut为C碱基在突变混池中的频率,wt为C碱基在野生型混池中的频率;mut为G碱基在突变混池中的频率,wt为G碱基在野生型混池中的频率;mut为T碱基在突变混池中的频率,wt为T碱基在野生型混池中的频率。
1.5 候选基因定位及分析
以Wm82.a2.v1 (Phytozome info:Wm82.a2.v1 (doe.gov))为参考基因组, 根据所得到的QTL区间, 对候选区段内的基因进行鉴定。利用基因功能注释等对候选基因进行功能注释。
1.6 候选基因实时荧光定量PCR
选取SIM-0 d、SIM-14 d、SEM-3 d、SEM-5 d、SEM-7 d、SEM-14 d (丛生芽诱导0 d、14 d; 丛生芽伸长3、5、7、14 d)共6个时期的亲本材料DN50、极端材料Z14及Z39, 进行RNA的提取, 提取步骤参照RNA提取试剂盒(OMEGA Plant RNA Kit (50), R6827-01)试剂盒)说明书进行。将提取的RNA反转录为单链cDNA, 试验步骤参照RNA反转录试剂盒(HiScript III 1st Strand cDNA Synthesis Kit (+gDNA wiper), (Vazyme R312-01)试剂盒)说明书进行。按照荧光定量PCR仪试剂盒(ChamQ Universal SYBR qPCR Master Mix)说明书进行检测分析。反应体系盒扩增程序参照荧光定量PCR试剂盒说明书进行, 每个样品进行3次重复, 相对表达量的计算采用2–ΔΔCt法[33]。
2 结果与分析
2.1 大豆再生种质筛选
对父母本器官发生能力进行试验(图2)发现, 在丛生芽诱导、丛生芽伸长及生根3个阶段, DN50生长情况明显好于Keburi, 在显微镜下可以明显观察到(图3) DN50的丛生芽饱满、密集且数量较多, Keburi的丛生芽生长较小、数量较少。根据RP、RA、RE、RR 4个指标统计分析数据(图4)发现, DN50在各个时期均显著高于Keburi。表明DN50再生较好; Keburi再生较弱。

图2 DN50和Keburi各个时期生长情况
A、D: 丛生芽诱导; B、E: 丛生芽伸长; C、F: 生根情况。标尺为1 cm。
A, D: adventitious shoots induction; B, E: adventitious shoots elongation; C, F: rooting situation. Bar: 1 cm.
红色三角形标注点处均为丛生芽。A: DN50; B: Keburi. The red triangles are marked with clustered buds.
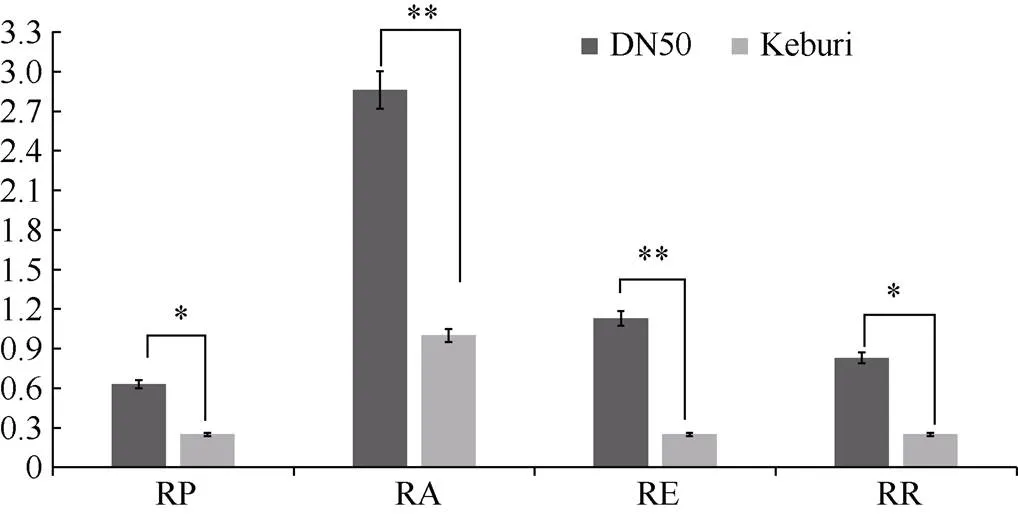
图4 父母本再生指标对比
RP: 再生潜力; RA: 再生能力; RE: 再生效率; RR: 再生率。值由假设方差相等的双尾检验, **< 0.01, *< 0.05。
RP: regenerative potential; RA: regenerative ability; RE: regenerative efficiency; RR: regenerative rate.-values are tested by a two-tailed-test assuming equal variances. **< 0.01, *< 0.05.
对子代200份材料进行试验(图5)发现, 再生较强材料在各个时期其生长好于再生较弱材料。在显微镜下可以观察到(图6), 再生较好材料丛生芽数目多、丛生芽生长密集较为明显, 而再生较差材料丛生芽聚集在小部分区域, 数目较少。
以RP、RA、RE、RR、主成分值、隶属函数值、值统计分析200份子代材料(图7), 最终根据值排序筛选到20份再生较强及20份再生较弱的材料作为极端材料。由表1可知, 2个主成分累计贡献率达到95.57%, 表明这2个主成分就可较好的反应最初4个单项指标所涵盖的大部分内容, 可通过2个主成分概括群体再生能力间的相关关系。由表2可知, 强再生材料值为1.88~6.55; 弱再生材料值为-1.03~-0.85。其中20份强再生材料分别为Z149、Z52、Z4、Z176、Z184、Z30、Z53、Z155、Z3、Z16、Z98、Z35、Z2、Z168、Z77、Z20、Z142、Z1、Z6、Z14, 其中Z14再生最强Z6次之; 20份弱再生材料分别为Z39、Z153、Z121、Z152、Z95、Z161、Z165、Z193、Z44、Z84、Z147、Z197、Z25、Z62、Z96、Z108、Z111、Z120、Z127、Z129。

图5 极端材料各个时期生长情况
a: 强再生材料; b: 弱再生材料。A、D: 丛生芽诱导; B、E: 丛生芽伸长; C、F: 生根情况。标尺为1 cm。
a: regenerative good materials; b: regenerative poor materials. A, D: adventitious shoots induction; B, E: adventitious shoots elongation; C, F: rooting situation. Bar: 1 cm.

图6 显微镜下极端材料丛生芽生长情况
A: 再生好材料; B: 再生差材料。红色三角形标注点处均为丛生芽。
A: regenerative good materials; B: regenerative poor materials. The red triangles are marked with clustered buds.
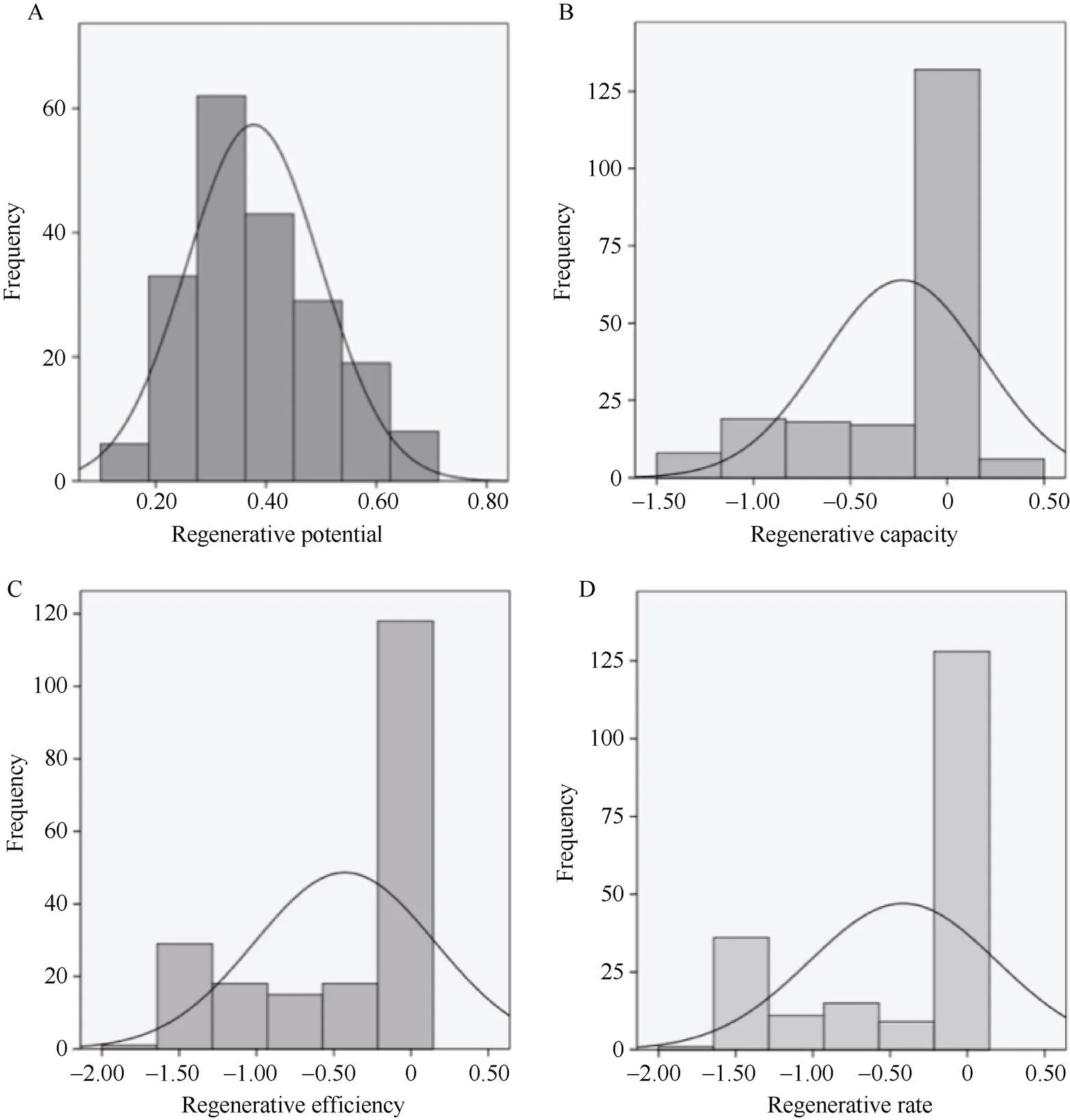
图7 子代材料数据统计
A: 再生潜力统计; B: 再生能力统计; C: 再生效率统计; D: 再生率统计。
A: the regenerative potential; B: the regenerative capacity; C: the regenerative efficiency; D: the regeneration rate.
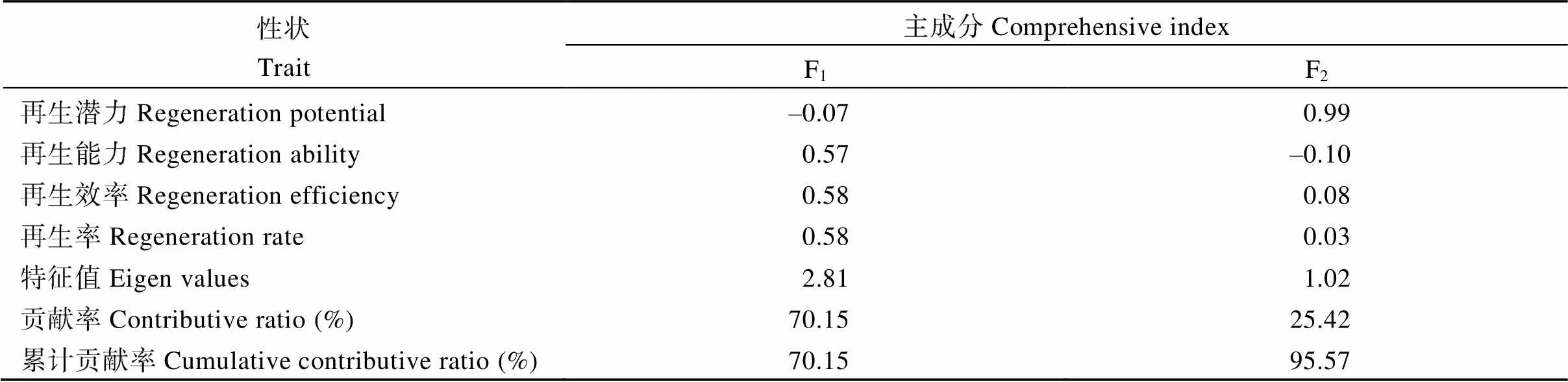
表1 主成分特征值及贡献率
2.2 BSA混池测序定位再生QTL
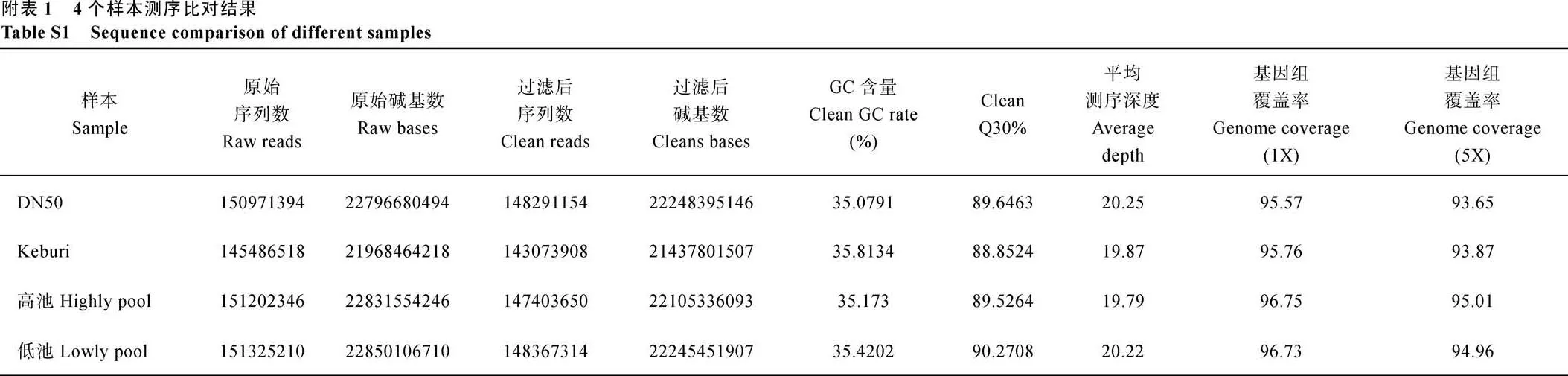
将筛选到的再生能力较强的20份材料和再生能力较弱的20份材料分别构建混合池, 对所有数据进行测序分析, 本次测序共获得88.04 G的clean data, Q30达到89.57%, 混池测序平均深度为20.03×。样品与参考基因组平均比对效率为99.15%, 4个样本1×覆盖率百分比95%以上, 5×覆盖率百分比93%以上(附表1), 对比对结果进行统计, 发现测序数据均有较好的覆盖深度和覆盖度, 参考基因组被均匀覆盖, 随机性良好, 可进行后续BSA-seq关联位点分析。
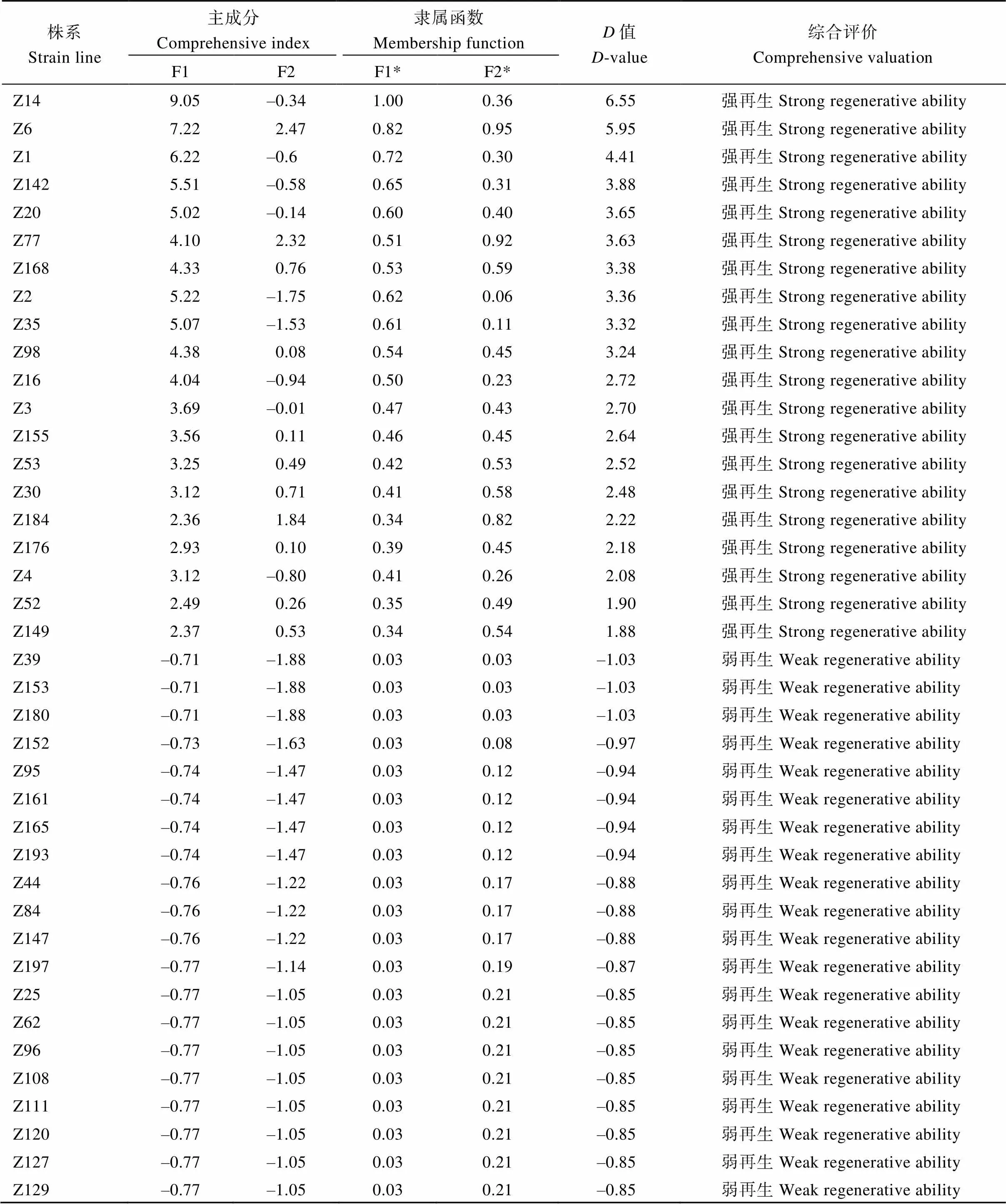
表2 极端材料的主成分值、隶属函数值、D值
F1, F2为主成分1, 成分2。F1*, F2*为隶属函数1, 隶属函数2。
F1, F2 are the main components 1 and 2. F1*, F2* are membership functions 1 and 2.
根据测序结果通过关联分析筛选候选区间, 采用ED值方法进行定位。ED关联结果如图8所示, 在18号染色体共得到2个区域, 总长度为2 Mb, 共包含379个基因(表3)。
2.3 关联区域基因注释富集分析
利用GO数据库对候选区间内的379个基因进行功能注释(图9), 富集结果显示, 注释基因主要参与的前20个条目主要包括植物型细胞壁组织或生物发生(GO: 0071669)、纤维素微纤维组织(GO: 0010215)、细胞外基质组织(GO: 0030198)、胞间连丝(GO: 0009506)、转录因子TFID复合物(GO: 0005669)等, 根据adjust值最小即为最显著富集, 本研究发现磷酸二酯水解酶活性、甲基转移酶活性、核DNA−定向RNA聚合酶复合物、植物型细胞壁组织或生物发生被显著富集, 其中研究发现植物型细胞壁组织或生物发生(GO: 0071669)与植物生长发育息息相关, 可能在植物再生过程中发挥关键作用。有6个基因被富集到此条目中, 分别为、、、、、, 因此本研究将这6个基因作为候选基因, 对其进行功能注释(附表2),含有Pollen allergen结构域, 拟南芥同源基因为, 在细胞壁发挥作用;为膨胀素相关基因, 拟南芥同源基因为, 该基因在细胞壁发挥作用;、均为COBRA家族基因, 拟南芥同源基因为, 基因在细胞膜中发挥作用;为COBRA-like蛋白家族成员, 拟南芥同源基因为, 该基因在细胞膜发挥作用;为UDP-ARABINOPYRANOSE MUTASE家族基因, 拟南芥同源基因为, 该基因在高尔基体中发挥作用。
2.4 候选基因表达模式分析
以3个品种DN50、Z14、Z39的6个时期(SIM-0、SIM-14、SEM-3、SEM-5、SEM-7、SEM-14)作为材料, DN50的表达量为对照, 分别对6个候选基因进行实时荧光定量分析(图10), 结果表明6个候选基因在再生强的材料中SEM-5时期表达量均较高, SEM-5为丛生芽伸长5 d, 再生强材料在此时期会产生大量伸长的丛生芽, 表达量较高推测是由于丛生芽伸长期间细胞膨胀、细胞壁伸长导致。再生弱材料在此时期表达量较低, 推测是由于再生弱材料在生长发育期间产生丛生芽较少。这些基因对大豆再生的影响将在后续的工作中进行验证。
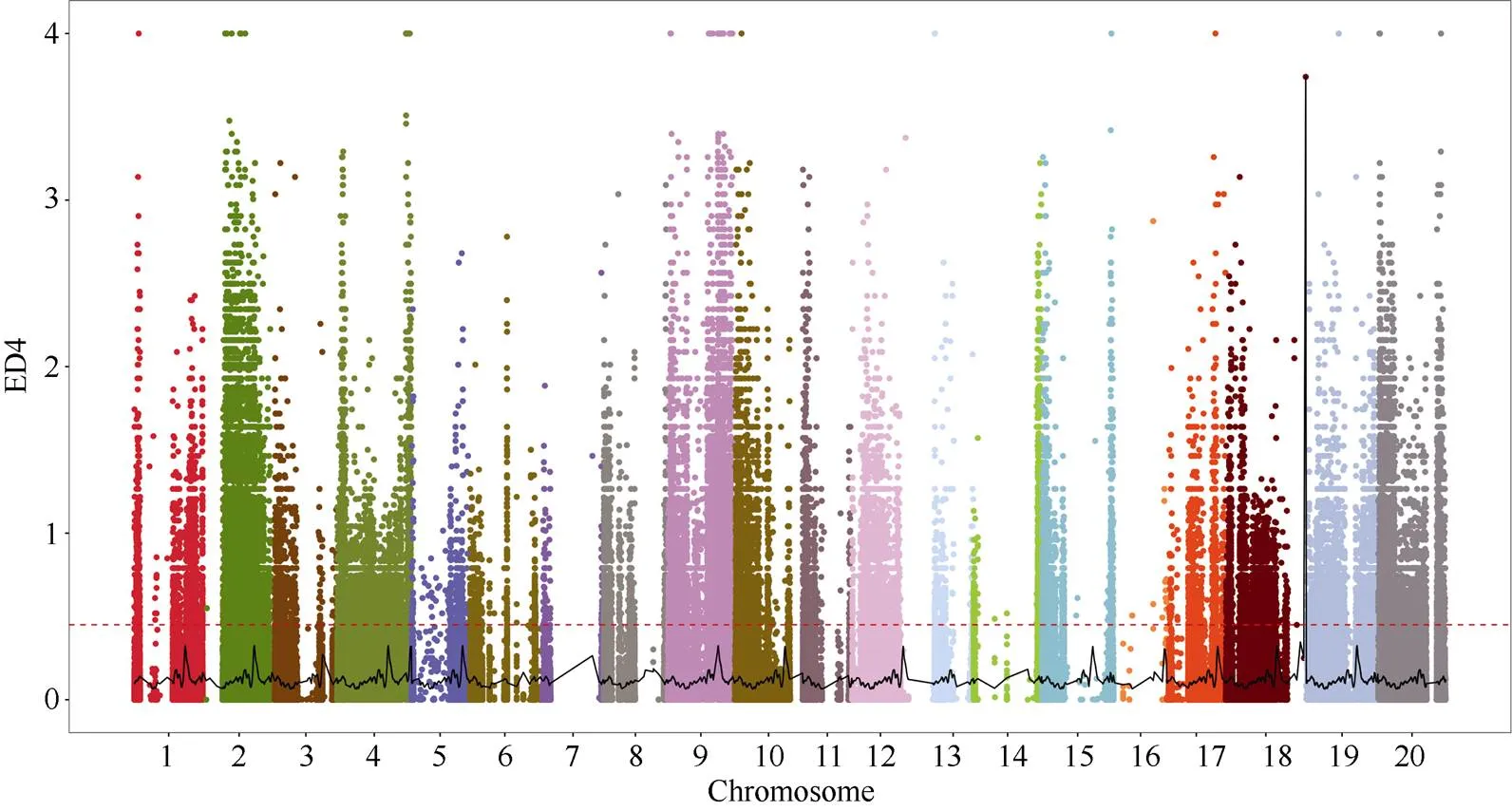
图8 ED关联值在染色体上的分布
横坐标为染色体名称, 彩色的点代表每个SNP位点的ED值, 黑色的线为拟合后的ED值, 红色的虚线代表显著性关联阈值, ED值越高, 代表该点关联效果越好。
The abscissa is the chromosome name; the colored dots represent the ED value of each SNP site; the black line is the ED value after fitting; the red dashed line represents the significance association threshold; the higher the ED value, the better the point association effect.
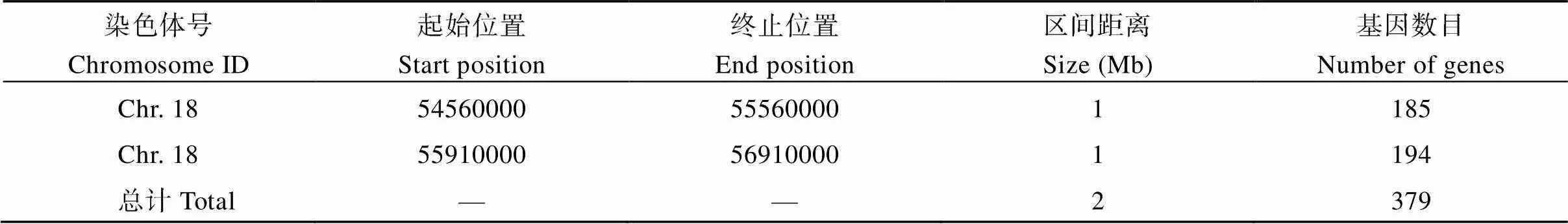
表3 关联分析基因区域内信息统计
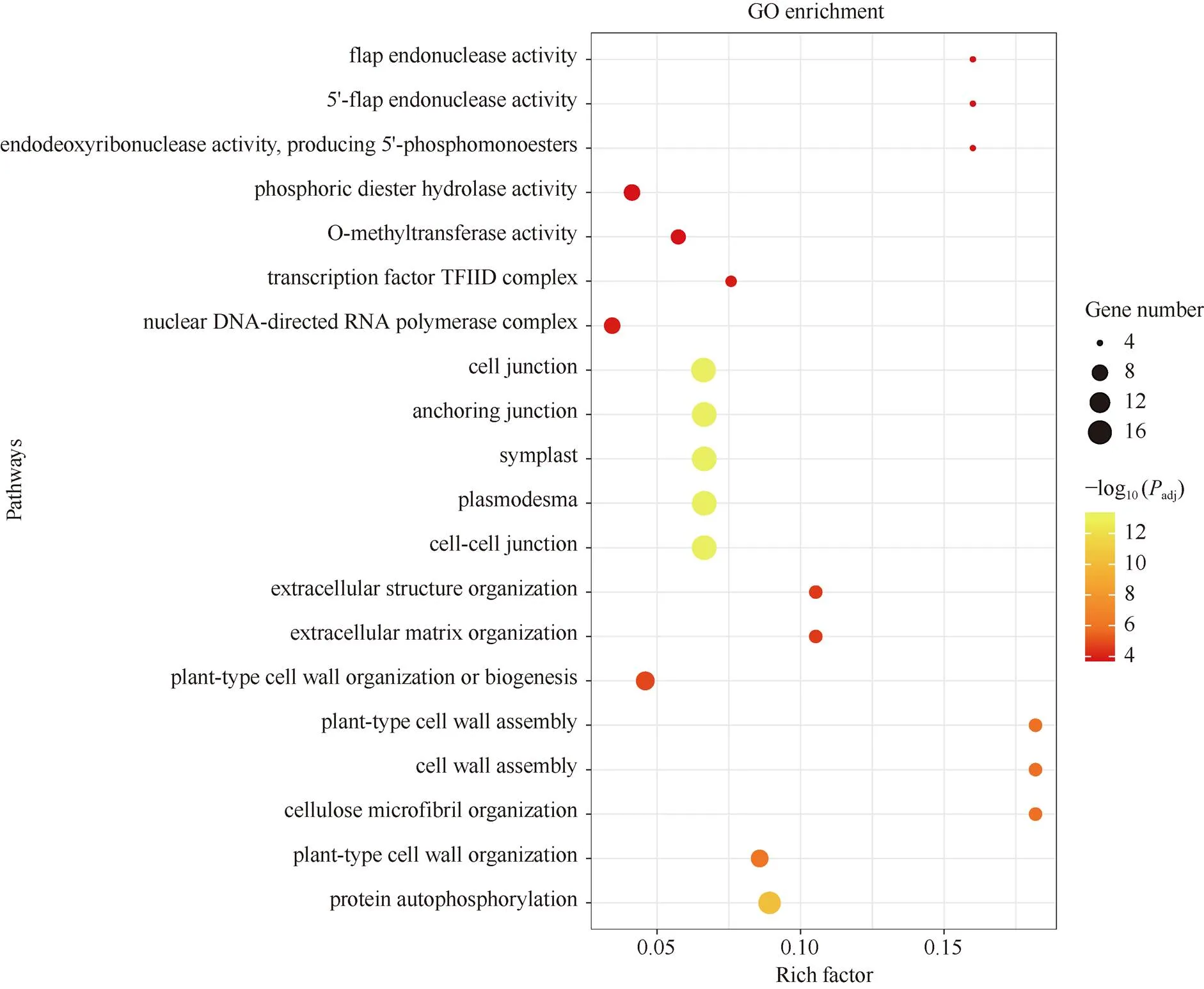
图9 候选区域内基因GO注释聚类图
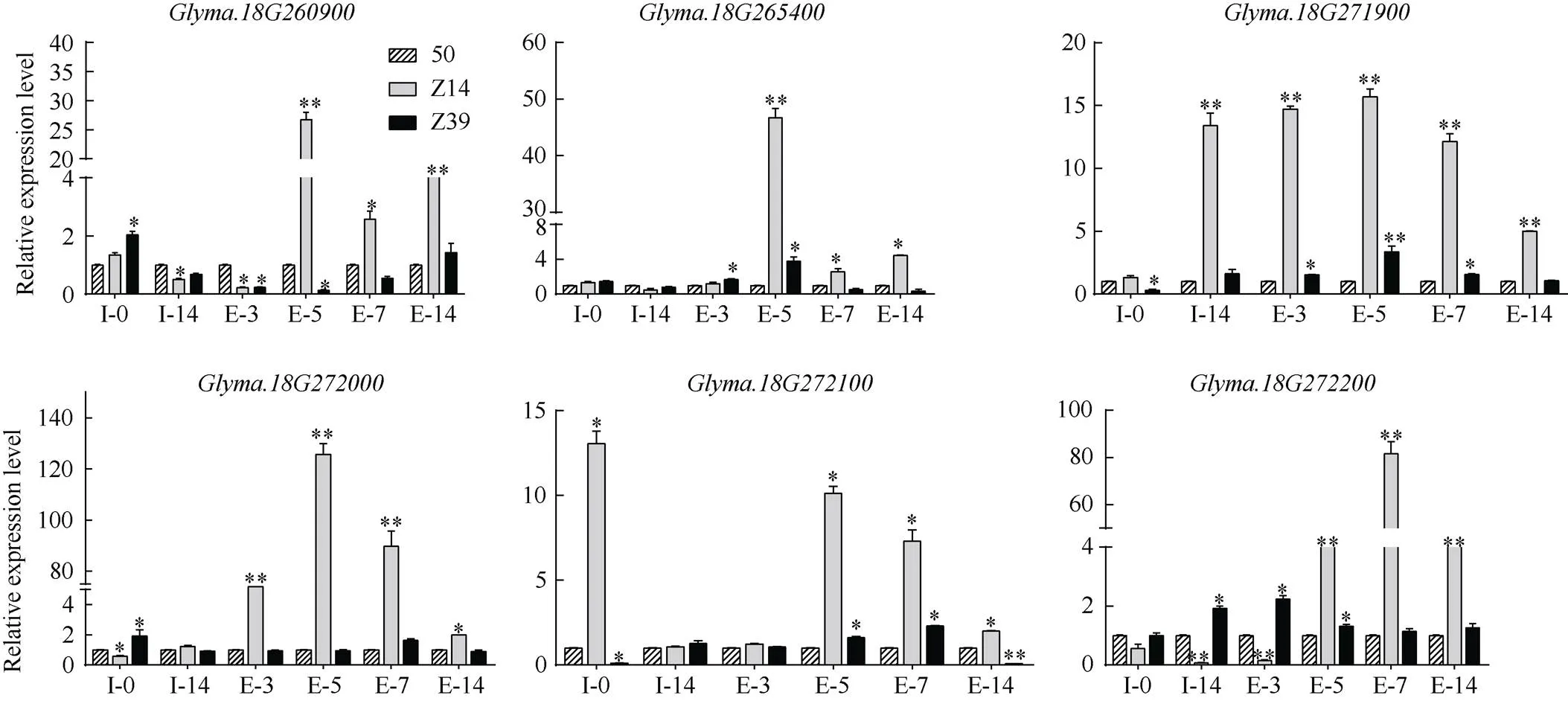
图10 候选基因在不同时期不同材料中的表达量
I-0为SIM-0; I-14为SIM-14; E-3为SEM-3; E-5为SEM-5; E-7为SEM-7; E-14为SEM-14。*和**分别在0.05和0.01概率水平差异显著。
I-0: SIM-0; I-14: SIM-14; E-3: SEM-3; E-5: SEM-5; E-7: SEM-7; E-14: SEM-14. * and ** indicate significantly different at the 0.05 and 0.01 probability levels, respectively.
3 讨论
3.1 不同基因型对大豆再生情况影响
目前, 主栽品种再生能力弱受到基因型的束缚, 且大豆组织培养对基因型有明显的依赖性[34], 并且大豆的转化率远低于其他双子叶植物拟南芥、烟草等。因为大豆基因型差异是影响大豆遗传转化的重要因素, 筛选分化能力强的基因型是大豆遗传转化的先决条件。大豆子叶节丛生芽诱导分化体系受基因型的限制, 不同基因型大豆由于遗传背景与生理的差异导致诱导分化频率具有很大的差异。曹劲宏等[35]研究47份不同基因型小豆胚尖再生能力, 筛选出9份再生能力较好的小豆资源; 武小霞等[36]对北方37个大豆品种进行了基因型筛选确定了5个基因型分化率较高; 滕卫丽等[37]利用子叶节再生体系筛选得到8个高再生率大豆品种, 为选育高再生率品种提供了基础材料; 李换丽等[38]试验发现晋豆37号和中黄13号在子叶节再生体系中再生能力明显高于其他品种。以上结果都是利用不同再生能力基因型的大豆为材料进行筛选得到的高再生率品种, 所以本研究利用东农50和Keburi及其衍生RILs为材料, 评价大豆再生潜力、再生能力、再生效率和再生率, 最终筛选得到强再生的家系分别为Z14和Z6, 其中Z14再生潜力高于母本DN50, Z6在再生能力上高于母本DN50, 这些材料可以作为选育高再生能力品种提供材料。
3.2 候选基因功能
目前, 对大豆再生基因的研究鲜有报道, 因此对提高大豆再生能力的基因进行挖掘, 丰富大豆再生的基因调控网络有重要影响。而植物再生能力主要与细胞的分裂与扩张有关, 本研究定位到候选区间内的6个候选基因可能与再生能力有关, 6个候选基因基本信息如表6所示,、、3个基因均为基因家族, 在Roudier等[39]在拟南芥中的研究表明,基因具有参与纤维素微纤维定向的功能, 这个功能是植物形态发生过程中异性膨胀的一个重要因素。Ko等[40]研究发现COBRA蛋白在根和芽组织的细胞结构的生长过程中起着重要作用。Anoop等[41]研究发现在玉米中COBRA家族成员在纤维素合成中发挥功能, 在次生细胞壁发育发挥作用。这些现象也印证了qRT-PCR实验中,基因在植物组织表达的结果。基因在植物形态发生过程中会起到扩张作用, 该蛋白编码糖基磷脂酰肌醇锚定蛋白, 在细胞壁扩张和纤维素沉积中发挥作用, 并且亚细胞定位预测在细胞壁, 由此推测该基因可能参与植物的再生, 并且我们推测该家族基因与植物根及芽生长相关, 可能提高植物的再生能力。基因含有Pollen-allerg-1结构域, 为扩展家族典型结构域, 在植物生长发育过程中诱导细胞壁松弛和伸展, 细胞壁松弛有助于发育过程中的细胞增大[42]。某些含有Pollen-allerg-1结构域的蛋白可以被生长素、赤霉素诱导表达[42], 并且该基因亚细胞定位预测在细胞壁中, 所以在大豆的发育过程中可能会诱导细胞壁的伸展, 促进细胞的增大使得在大豆的再生过程中发挥作用。为膨胀素相关基因, 许多研究表明, 植物从发芽到结果几乎所有的生理发育过程都需要膨胀素[43-44], 该基因亚细胞定位预测也在细胞壁中, 在大豆发芽、伸长到生根过程中使细胞膨胀增大, 促进丛生芽的生长、苗的伸长, 参与植物再生过程中的生长发育。为()家族基因,与发育等重要的生物过程相关[45-46], 在水稻中在植物发育过程中也普遍表达, 有些主要在生殖组织中表达[47], 并且该基因亚细胞定位预测在高尔基体, 高尔基体可以合成果胶和非纤维素多糖, 与细胞壁的形成有关, 在大豆再生过程中可能起到促进伸长苗伸长, 对大豆发育过程发挥一定的作用。这些基因对大豆再生的影响将在后续的试验中进行验证。
4 结论
采用器官发生试验筛选出极端材料各20份, 基于BSA-seq技术将与大豆再生相关基因初步定位到18号染色体上的2个区域内, 最终筛选出6个可能与大豆再生相关的候选基因。本试验为再生新品种选育提供参考和重要材料; 为挖掘大豆再生基因、探索大豆再生规律、提高大豆遗传转化效率奠定一定理论基础。
[1] Cheng T Y, Saka H, Voqui-dinh T H. Plant regeneration from soybean cotyledonary node segments in culture., 1980, 19: 91–99.
[2] Wright M S, Ward D V, Hinchee M A, Carnes M G, Kaufman R J. Regeneration of soybean (L. Merr.) from cultured primary leaf tissue., 1987, 6: 83–89.
[3] Barwale U B, Kerns H R, Widholm J M. Plant regeneration from callus cultures of several soybean genotypes via embryogenesis and organogenesis., 1986, 167: 473–481.
[4] Kim J, Lamotte C E, Hack E. Plant regeneration in vitro from primary leaf nodes of soybean () seedlings., 1990, 136: 664–669.
[5] 武小霞, 孙晶, 苏安玉, 李静, 刘明, 张彬彬, 李冬梅, 李文滨. 大豆再生抑制消减杂交文库的构建. 东北农业大学学报, 2014, 45(7): 38–44. Wu X X, Sun J, Su A Y, Li J, Liu M, Zhang B B, Li D M, Li W B. Soybean regeneration inhibits the construction of a subtractive hybrid library., 2014, 45(7): 38–44 (in Chinese with English abstract).
[6] Kartha K K, Pahl K P, Leung N L, Mroginski L A J B. Plant regeneration from meristems of grain legumes: soybean, cowpea, peanut, chickpea, and bean., 1981, 59: 1671–1679.
[7] Paz M M, Martinez J C, Kalvig A B, Fonger T M, Wang K. Improved cotyledonary node method using an alternative explant derived from mature seed for efficientmediated soybean transformation., 2006, 25: 206–213.
[8] 王怡婷, 赵莹, 李思琪, 于耸, 郑志民. 大豆U6启动子(YT9)在CRISPR/Cas9基因组编辑体系中的功能分析. 分子植物育种, 2023, 21: 1189–1195. Wang Y T, Zhao Y, Li S Q, Yu S, Zheng Z M. Functional analysis of the soybean U6 promoter (YT9) in CRISPR/Cas9 genome editing systems., 2023, 21: 1189–1195 (in Chinese with English abstract).
[9] Li S, Cong Y, Liu Y, Wang T, Shuai Q, Chen N, Gai J, Li Y. Optimization of agrobacterium-mediated transformation in soybean., 2017, 8: 246.
[10] Fletcher J C, Brand U, Running M P, Simon R, Meyerowitz E M. Signaling of cell fate decisions by CLAVATA3 inshoot meristems., 1999, 283: 1911–1914.
[11] Long J A, Moan E I, Medford J I, Barton M K. A member of the KNOTTED class of homeodomain proteins encoded by the STM gene of., 1996, 379: 66–69.
[12] Endrizzi K, Moussian B, Haecker A, Levin J Z, Laux T. The SHOOT MERISTEMLESS gene is required for maintenance of undifferentiated cells inshoot and floral meristems and acts at a different regulatory level than the meristem genes WUSCHEL and ZWILLE., 1996, 10: 967–979.
[13] Zuo J, Niu Q W, Frugis G, Chua N H. Thegene promotes vegetative-to-embryonic transition in., 2002, 30: 349–359.
[14] Leibfried A, To J P, Busch W, Stehling S, Kehle A, Demar M, Kieber J J, Lohmann J U. WUSCHEL controls meristem function by direct regulation of cytokinin-inducible response regulators., 2005, 438: 1172–1175.
[15] Mayer K F, Schoof H, Haecker A, Lenhard M, Jürgens G, Laux T. Role of WUSCHEL in regulating stem cell fate in theshoot meristem., 1998, 95: 805–815.
[16] Su Y H, Zhou C, Li Y J, Yu Y, Tang L P, Zhang W J, Yao W J, Huang R, Laux T, Zhang X S. Integration of pluripotency pathways regulates stem cell maintenance in theshoot meristem., 2020, 117: 22561–22571.
[17] Matsuo N, Makino M, Banno H.ENHANCER OF SHOOT REGENERATION (ESR)1 and ESR2 regulateshoot regeneration and their expressions are differentially regulated., 2011, 181: 39–46.
[18] Banno H, Ikeda Y, Niu Q W, Chua N H. Overexpression ofESR1 induces initiation of shoot regeneration., 2001 13: 2609–2618.
[19] 蔡英卿. 龙眼体胚发生过程中SERK等胚性相关基因的克隆与表达分析. 福建农林大学博士学位论文, 福建福州, 2011. Cai Y Q. Cloning and Expression Analysis of Embryonic Genes Such as SERK during Longan Embryogenesis. PhD Dissertation of Fujian Agriculture and Forestry University, Fuzhou, Fujian, China, 2011 (in Chinese with English abstract).
[20] Lutz K A, Martin C, Khairzada S, Maliga P. Steroid-inducible BABY BOOM system for development of fertileplants after prolonged tissue culture., 2015, 34: 1849–1856.
[21] Horstman A, Li M, Heidmann I, Weemen M, Chen B, Muino J M, Angenent G C, Boutilier K. The BABY BOOM transcription factor activates the LEC1-ABI3-FUS3-LEC2 network to induce somatic embryogenesis., 2017, 175: 848–857.
[22] Li S N, Cheng P, Bai Y Q, Shi Y, Yu J Y, Li R C, Zhou R N, Zhang Z G, Wu X X, Chen Q S. Analysis of soybean somatic embryogenesis using chromosome segment substitution lines and transcriptome sequencing.(Basel), 2019, 10: 943.
[23] 曾维英, 苏燕竹, 赖振光, 杨守臻, 陈怀珠, 谭玉荣, 孙祖东, 盖钧镒. 基于BSA-Seq技术鉴定大豆耐荫性状相关候选基因. 中国油料作物学报, 2021, 43: 1006–1015. Zeng W Y, Su Y Z, Lai Z G, Yang S Z, Chen H Z, Tan Y R, Sun Z D, Gai J Y. Identification of candidate gene controlling shade- tolerant by BSA-Seq in soybean., 2021, 43: 1006–1015 (in Chinese with English abstract).
[24] 张之昊, 王俊, 刘章雄, 邱丽娟. 基于BSA-Seq技术挖掘大豆中黄622的多小叶基因. 作物学报, 2020, 46: 1839–1849. Zhang Z H, Wang J, Liu Z X, Qiu L J. Mapping of an incomplete dominant gene controlling multifoliolate leaf by BSA-seq in soybean (L.)., 2020, 46: 1839–1849 (in Chinese with English abstract).
[25] 严昕, 项超, 刘荣, 李冠, 李孟伟, 李正丽, 宗绪晓, 杨涛. 基于BSA-seq技术对豌豆花色基因的精细定位. 作物学报, 2023, 49: 1006–1015. Yan X, Xiang C, Liu R, Li G, Li M W, Li Z L, Zong X X, Yang T, Fine mapping of flower colour gene in pea (L.) based on BSA-seq technique., 2023, 49: 1006–1015 (in Chinese with English abstract).
[26] Zhong C, Sun S, Li Y, Duan C, Zhu Z. Next-generation sequencing to identify candidate genes and develop diagnostic markers for a novel Phytophthora resistance gene,, in soybean., 2018, 131: 525–538.
[27] 曾维英, 赖振光, 孙祖东, 杨守臻, 陈怀珠, 唐向民. 基于BSA-Seq和RNA-Seq方法鉴定大豆抗豆卷叶螟候选基因. 作物学报, 2021, 47: 1460–1471.Zeng W Y, Lai Z G, Sun Z D, Yang S Z, Chen H Z, Tang X M. Identification of the candidate genes of soybean resistance to bean pyralid (Fabricius) by BSA-Seq and RNA-Seq., 2021, 47: 1460–1471 (in Chinese with English abstract).
[28] 李毅丰. 番茄节间长度主效基因定位及候选基因生物信息学分析. 河北科技师范学院硕士学位论文, 河北秦皇岛, 2022. Li Y F. Mapping of Major Genes and Bioinformatics Analysis of Candidate Genes in Tomato Internode Length. MS Thesis of Hebei Normal University of Science & Technology, Qinhuangdao, Hebei, China, 2022 (in Chinese with English abstract).
[29] 蒋家焕, 朱永生, 陈丽萍, 郑燕梅, 蔡秋华, 谢华安, 王爱荣, 张建福. 利用BSA-Seq方法定位一个水稻早衰相关基因. 福建农业学报, 2022, 37(2): 131–137. Jiang J H, Zhu Y S, Chen L P, Zheng Y M, Cai Q H, Xie A H, Wang A R, Zhang J F. Mapping of early senescence-relatedin rice by BSA-seq technique., 2022, 37(2): 131–137 (in Chinese with English abstract).
[30] 王雪彬, 张健, 韦燕燕, 罗继景, 梁云涛, 蔡中全. 基于BSA-seq的水稻耐陈化QTL定位分析. 分子植物育种, 2021. 2021-11-05. https://kns.cnki.net/kcms/detail/46.1068.S.20211104. 1822.015.html. Wang X B, Zhang J, Wei Y Y, Luo J J, Liang Y T, Cai Z Q. QTLs Mapping on rice grain aging tolerance based on BSA-seq., 2021. 2021-11-05. https://kns.cnki.net/kcms/detail/ 46.1068.S.20211104.1822.015.html (in Chinese with English abstract).
[31] 周雨晴, 郭宇玲, 伊然, 陈壁融, 杨羽清, 潘玉朋. 基于BSA-Seq的黄瓜重要园艺性状遗传定位研究进展. 分子植物育种, 2022. 2022-07-05. https://kns.cnki.net/kcms/detail/46.1068. S.20220704.0904.002.html. Zhou Y Q, Guo Y L, Yi R, Chen B R, Yang Y Q, Pan Y P. Research progress on genetic mapping of cucumber important horticultural traits based on BSA-Seq., 2022. 2022-07-05. https://kns.cnki.net/kcms/detail/46.1068.S.20220704. 0904.002.html (in Chinese with English abstract).
[32] Hill J T, Demarest B L, Bisgrove B W, Gorsi B, Su Y C, Yost H J. MMAPPR: mutation mapping analysis pipeline for pooled RNA-seq., 2013, 23: 687–697.
[33] Livak K J, Schmittgen T D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method., 2001, 25: 402–408.
[34] Omatsuda T, Ohyama K. Genotypes of high competence for somatic embryogenesis and plant regeneration in soybean., 2004 75: 695–700.
[35] 曹劲宏, 牟少萌, 何雪, 赵波, 杨凯, 李奕松, 李玮瑜, 万平. 不同基因型小豆胚尖再生能力筛选和培养条件优化. 分子植物育种, 2015, 13: 106–118.Cao J H, Mou S M, He X, Zhao B, Yang K, Li Y S, Li W Y, Wan P. Screening on regeneration capacity of embryonic tip from different genotypes of adzuki bean () and optimizing on culture condition., 2015 13: 106–118 (in Chinese with English abstract).
[36] 武小霞, 李静, 姜成涛, 刘伟婷, 刘淼, 李文滨. 大豆子叶节再生中植物生长调节剂浓度及基因型筛选. 中国油料作物学报, 2011, 33: 123–129.Wu X X, Li J, Jiang C T, Liu W T, Liu M, Li W B. Optimization of regeneration system from soybean cotyledonary node., 2011, 33: 123–129 (in Chinese with English abstract).
[37] 滕卫丽, 郑立娜, 张琦, 赵雪, 韩英鹏, 李文滨. 大豆再生相关性状QTL定位. 东北农业大学学报, 2021, 52(4): 1–10. Teng W L, Zheng L N, Zhang Q, Zhao X, Han Y P, Li W B. QTL mapping of traits correlated with regeneration in soybean., 2021, 52(4): 1–10 (in Chinese with English abstract).
[38] 李换丽, 王丹, 张树伟, 雷佳, 吴霞, 王新胜, 马燕斌. 山西不同大豆品种再生体系的筛选. 山西农业科学, 2020, 48(2): 161–166. Li H L, Wang D, Zhang S W, Lei J, Wu X, Wang X S, Ma Y B. Selection of soybean regeneration system with different verieties in Shanxi., 2020, 48(2): 161–166 (in Chinese with English abstract).
[39] Roudier F O, Fernandez A G, Fujita M, Himmelspach R, Borner G H H, Schindelman G, Song S, Baskin T I, Dupree P, Wasteneys G O, Benfey P N. COBRA, anextracellular glycosyl-phosphatidyl inositol-anchored protein, specifically controls highly anisotropic expansion through its involvement in cellulose microfibril orientation., 2005, 17: 1749–1763.
[40] Ko J H, Kim J H, Jayanty S S, Howe G A, Han K H. Loss of function of COBRA, a determinant of oriented cell expansion, invokes cellular defence responses in., 2006, 57: 2923–2936.
[41] Sindhu A, Langewisch T, Olek A, Multani D S, McCann M C, Vermerris W, Carpita N C, Johal G. Maize Brittle stalk2 encodes a COBRA-like protein expressed in early organ development but required for tissue flexibility at maturity., 2007, 145: 1444–1459.
[42] Zhang W, Yan H, Chen W, Liu J, Jiang C, Jiang H, Zhu S, Cheng B. Genome-wide identification and characterization of maize expansin genes expressed in endosperm., 2014, 289: 1061–1074.
[43] Marowa P, Ding A, Kong Y. Expansins: roles in plant growth and potential applications in crop improvement., 2016, 35: 949–965.
[44] Fleming A J, McQueen-mason S J, Mandel T, Kuhlemeier C J S. Induction of leaf primordia by the cell wall protein expansin., 1997, 276: 1415–1418.
[45] Zavaliev R, Sagi G, Gera A, Epel B L. The constitutive expression ofplasmodesmal-associated class 1 reversibly glycosylated polypeptide impairs plant development and virus spread., 2009, 61: 131–142.
[46] Gallardo K, Le Signor C, Vandekerckhove J, Thompson R D, Burstin J. Proteomics ofseed development establishes the time frame of diverse metabolic processes related to reserve accumulation., 2003, 133: 664–682.
[47] Sumiyoshi M, Inamura T, Nakamura A, Aohara T, Ishii T, Satoh S, Iwai H. UDP-arabinopyranose mutase 3 is required for pollen wall morphogenesis in rice ()., 2014, 56: 232–241.
Mining candidate genes related to soybean regeneration based on BSA-seq method
ZHAO Yu-Jing1,**, ZHANG Bin-Shuo1,**, SU An-Yu2,YU Zhen-Hai1, LI Jia-Huan1, LIN Yang1, ZHANG Yan-Ting1, WU Xiao-Xia1,*, and ZHAO Ying1,*
1College of Agronomy, Northeast Agricultural University, Harbin 150030, Heilongjiang, China;2College of Resources and Environment, Northeast Agricultural University, Harbin 150030, Heilongjiang, China
Transgenic breeding technology can improve soybean directionally, and provide a new idea for improving soybean yield. In order to search for the genes related to soybean regeneration, explore the rules of soybean regeneration, and improve the efficiency of genetic transformation, we conducted soybean organogenesis experiment with 200 materials including DN50 (a material with strong regeneration ability), Keburi (a material with weak regeneration ability), and RILs of its offspring with strong regeneration ability. Compared the differences in regeneration ability between different genotypes, 20 extreme materials each were screened. Preliminary localization of soybean regeneration candidate genes by BSA-seq (bulked segregant analysis sequencing) technology, 88.04 G clean data were obtained in the 2 Mb interval with an average sequencing depth of 20.03 ×. The differentially expressed genes were mainly enriched in 20 items such as cellulose microfiber tissue, plant type cell wall tissue, or biogenesis, among which there were 6 genes in plant type cell wall tissue or biogenesis item significantly enriched. The tissue expression analysis of 6 genes showed that the relative expression level was high during cluster bud elongation, which indicating that it played a role in the process of soybean regeneration and might be the key gene affecting soybean regeneration. This study provides the basic materials for breeding new regenerated soybean varieties, and confirms the feasibility of BSA-seq technology in mining regenerated genes.
soybean; regeneration; organogenesis; BSA-seq
10.3724/SP.J.1006.2023.24276
本研究由国家自然科学基金项目(31971899, 32272093), 国家重点研发计划项目(2021YFD120160204, 2021YFD12011040202)和黑龙江省自然科学基金项目(TD2022C003, JJ2022YX0475)资助。
This study was supported by the National Natural Science Foundation of China (31971899, 32272093), the National Key Research and Development Program of China (2021YFD120160204, 2021YFD12011040202), and the Heilongjiang Natural Science Foundation (TD2022C003, JJ2022YX0475).
武小霞, E-mail: xxwu2012@126.com; 赵莹, E-mail: tianshi198937@126.com
**同等贡献(Contributed equally to this work)
赵宇晶, E-mail: yujingzhao99@163.com
2022-12-12;
2023-04-17;
2023-05-12.
URL: https://kns.cnki.net/kcms/detail/11.1809.S.20230511.1818.002.html
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/).
猜你喜欢
今日农业(2022年16期)2022-11-09
中国化肥信息(2022年5期)2022-08-30
今日农业(2021年20期)2021-11-26
今日农业(2021年14期)2021-10-14
江河文学(2020年6期)2020-01-04
东坡赤壁诗词(2018年3期)2018-07-16
浙江农业科学(2016年11期)2016-05-04
现代检验医学杂志(2015年6期)2015-02-06
实验动物与比较医学(2014年5期)2014-02-28
读书(2014年8期)2014-01-19
