
新暗色柱节孢H1全基因组测序和注释
2023-09-12 00:57赵晓珍王红林马玉华郑乾明
西南农业学报 2023年7期
赵晓珍,齐 勇,王红林,马玉华,郑乾明
(贵州省农业科学院果树科学研究所,贵阳 550006)
【研究意义】火龙果(Hylocereusspp.Britt.& Rose)属仙人掌科(Cactaceae)量天尺属(Hylocereus)多年生果树,又名红龙果、青龙果或仙蜜果等。火龙果原产于中南美洲[1],目前在马来西亚、越南和中国等的热带和亚热带地区种植,在我国主要分布在海南、广西、广东、台湾、贵州和云南等地。目前,国内外报道较多的火龙果真菌病害主要有溃疡病[2-8]、炭疽病[9-10]等。溃疡病是最严重的病害之一,主要危害植株的茎和果实,严重降低果实商品价值。探讨火龙果溃疡病病原的基因组信息,有助于了解其遗传变异和感染机制,从而制定有效的防控策略和培育抗性品种。【前人研究进展】火龙果受溃疡病侵染后,发病初期在茎和果实表面形成褪绿小斑点,严重时病斑连成片,后期病斑突起;湿度大时病斑边缘形成水渍状,不断扩大和腐烂,果实外观斑驳。近年来,火龙果溃疡病在马来西亚[2]、以色列[3]、美国佛罗里达州[4]等和中国台湾[5]、广东[6]、海南[7]及广西[11]等地均有发生。该病在广西和海南的发病率分别为50%以上和100%,在美国佛罗里达州发病率达70%以上[4,7,11]。自2012年在台湾首次报道引起火龙果溃疡病的病原为新暗色柱节孢(Neoscytalidiumdimidiatum)[5]以来,我国广东、广西和海南等地及马来西亚、以色列和美国等也陆续报道由新暗色柱节孢引起的火龙果溃疡病[2-4,6-7]。新暗色柱节孢属葡萄座腔菌科(Botryosphaeriaceae),其异名包括Fusicoccumdimidiatum、Hendersonulatoruloides及Scytalidiumdimidiatum[12],可引起兰花叶斑病[13]和李、杏枯萎病[14-15]等植物病害。除侵染植物外,新暗色柱节孢还引起皮肤病和鼻窦炎等人类疾病[16-17]。近年来,以Pacbio和Nanopore平台为代表的单分子实时测序技术被广泛用于基因组测序和组装[18-20]。Illumina具有测序短读长和高准确率等优点[19],有学者分别采用Pacbio+Illumina混合策略对金针菇(Flammulinafiliformis)[21]、木贼镰孢菌(Fusariumequiseti)D25-1[22]、菜豆壳球孢(M.phaseolina)11-12和Al-1[23]、葡萄座腔菌(Botryosphaeriadothidea)sdau11-99[24]和Nanopore+Illumina混合策略对Laburnicolarhizohalophila[25]和尖孢镰刀菌(Fusariumoxysporum)[26]进行研究,均获得高质量的参考基因组序列。【本研究切入点】关于新暗色柱节孢的致病机制目前鲜有研究,也缺乏高质量的参考基因组;且尚无分离自火龙果的新暗色柱节孢基因组序列报道。【拟解决的关键问题】利用前期分离获得的火龙果溃疡病病原菌H1菌株,经纯培养、形态学和分子生物学鉴定为新暗色柱节孢。采用Nanopore PromethION+ Illumina NovaSeq混合策略对火龙果溃疡病病原菌新暗色柱节孢H1菌株开展基因组测序、组装和注释分析,为了解其基因组构成、遗传变异、致病机理和防控策略奠定基础。
1 材料与方法
1.1 材料
1.1.1 菌株 火龙果溃疡病病原菌H1菌株分离自贵州省镇宁县种植的火龙果品种黔蜜龙植株茎部,经鉴定为新暗色柱节孢(N.dimidiatum)[10]。
1.1.2 试剂 文库构建试剂盒(Nextera DNA Flex Library Prep,Illumina,USA),连接试剂盒(SQK-LSK109,Oxford Nanopore Technologies,UK)。
1.1.3 仪器设备 超声波破碎仪(M220,Covaris,USA),荧光计(Qubit 2.0,Life Technologies,USA),生物分析仪(Agilent 2100,Agilent Technologies,USA),全自动核酸剪切仪(Megaruptor 2,Diagenode,Belgium),全自动核酸回收仪(BluePippin,Sage Science,USA),Oxford Nanopore测序仪(PromethION,Oxford Nanopore Technologies UK),Illumina NovaSeq平台(Illumina,San Diego,CA,USA)。
1.1.4 软件 GenomeScope分析平台(https://github.com/schatzlab/genomescope),NECAT软件(https://github.com/xiaochuanle/NECAT),Racon软件(v.1.4.11,https://github.com/isovic/racon),Pilon软件(v.1.23,https:// github.com/broadinstitute/pilon),purge_haplogs软件(https://github.com/skingan/purge_haplotigs_multiBAM),BWA软件(v.0.7.17,https://github.com/lh3/bwa),BUSCO软件(v.3.0.1,https://github.com/RoyNexus/busco),RepeatModeler软件(v.1.0.4,https://github.com/rmhubley/RepeatModeler),RepeatMasker软件(v.4.0.5,http://www.repeatmasker.org/),BRAKER软件(v.2.1.4,https://github.com/GaiusLatestVersionAugustus/ BRAKER)。
1.2 方法
1.2.1 菌株培养和基因组DNA提取 将H1菌株接种于PDA培养基,28 ℃培养5 d。收集菌丝后液氮速冻,采用CTAB法[27]提取基因组DNA。
1.2.2 Illumina测序和K-mer分析 基因组DNA样品使用超声波破碎仪处理,然后使用文库构建试剂盒构建长度为150 bp的小片段插入文库。测序文库使用荧光计定量并稀释,再使用生物分析仪检测。合格文库使用Illumina NovaSeq平台测序。获得Raw reads后,过滤低质量的序列。过滤标准如下:去除不确定碱基含量超过5%的Raw reads;去除质量值不大于5的碱基含量达50%的Raw reads;去除含有接头污染的Raw reads;去除PCR扩增过程中产生的重复序列。过滤后的高质量序列使用GenomeScope进行K-mer分析,以估算基因组的大小、杂合率和重复片段含量等信息。
1.2.3 Nanopore测序 基因组DNA样品使用全自动核酸剪切仪随机打断,磁珠富集和纯化大片段DNA。使用全自动核酸回收仪回收大片段,进行损伤修复、纯化、末端修复并加A。利用连接试剂盒连接,使用荧光计定量。测序文库加入R9.4流动槽,转移到PromethION测序仪测序。获得原始序列后,过滤去除平均质量值≤7的序列。以上实验操作委托武汉贝纳科技服务有限公司开展。
1.2.4 基因组组装 首先利用Nanopore PromethION平台测序获得的高质量序列,使用NECAT软件进行序列纠错和基因组组装,获得初步组装结果。然后使用Racon软件结合Nanopore数据对拼接结果进行2轮自我纠错;再使用Pilon软件对Illumina数据进行2轮纠错。最后使用purge_haplogs去除杂合获得组装结果。使用BWA程序将Illumina数据比对到基因组。使用BUSCO程序选取真核和真菌直系同源基因评估完整性。
1.2.5 基因组注释 (1)重复序列预测。通过TRF软件预测串联重复序列;使用RepeatModeler软件重复序列文库;结合repbase文库,使用RepeatMasker软件预测基因组中的转座子。
(2)非编码RNA预测。使用tRNAscan-SE(v.1.23)和RNAmmer(v.1.2)分别预测tRNA和rRNA,再使用Infernal程序(v.1.1.1)预测miRNA和snRNA。
(3)编码基因预测和注释。基因组的编码基因预测使用BRAKER软件,选择“Fungus”选项。利用diamond blastp程序(v.2.9.0,e值=1e-5)在Uniprot、Refseq、NR、COG、GO和KEGG数据库进行序列相似性检索。将KEGG注释获得的结果,使用KOBAS程序关联到KEGG orthology以及PATHWAY。基于Uniprot数据库中的Swiss-Port结果关联性,获得eggNOG数据库的注释信息,然后挑选COG注释结果分类。使用Hmmscan(v.3.1;e值=1e-2)在Pfam数据库和InterProScan数据库进行Motif查找和结构域预测。
2 结果与分析
2.1 Illumina测序及K-mer分析
从表1可知,通过Illumina NovaSeq平台对N.dimidiatumH1测序共获得6.04 Gb原始数据。去除低质量的序列,共获得6.03 Gb纯净数据。对纯净数据开展K-mer分析,N.dimidiatumH1基因组规模约45.06 Mb,杂合率为0.079%。
2.2 Nanopore测序和基因组组装
Nanopore PromethION平台测序获得7.93 Gb原始数据,序列数量为639 418条。进一步过滤低质量序列,获得7.67 Gb纯净数据,序列数量为614 580条。平均长度为12.49 Kb,最大长度为156.40 Kb,N50长度为23.19 Kb,平均质量值为10.25。
基于Nanopore平台获得的纯净数据进行基因组组装,并利用Illumina平台的纯净数据纠错再去杂合,最终获得的N.dimidiatumH1基因组大小为43.78 Mb,测序深度为175×。contig数量为14条,Gap数量为0。N50长度为3.92 Mb,平均长度为3.13 Mb,最小长度为175.69 Kb,最大长度为5.63 Mb。
2.3 组装完整性评估
使用Pilon软件将Illumina测序数据比对到组装的N.dimidiatumH1基因组序列上,其比对重复率为98.86%,覆盖率为99.94%。从表2看出,在真核基因集290个直系同源基因中,完整的单拷贝基因为286个(98.6%),完整的多拷贝基因为1个(0.3%),缺失基因仅3个(1%)。在真菌基因集758个直系同源基因中,完整的单拷贝基因为747个(98.5%),完整的单拷贝基因为2个(0.3%),基因片段仅2个(0.3%),缺失基因仅7个(0.9%)。上述结果表明,组装获得的N.dimidiatumH1基因组完整性较好。
2.4 基因组的结构预测
2.4.1 编码基因 对N.dimidiatumH1基因组中编码基因预测表明,编码蛋白的基因共有12 962个,其mRNA平均长度为1800 bp,绝大部分长度分布于300~9000 bp,以600~2100和6000~9000 bp基因较多(图1)。编码基因的序列(CDS)平均长度为1541.34 bp,绝大部分长度分布于300~6000 bp,以长度300~1800 bp的基因数量较多。外显子数量共计41 149个,平均长度为485.52 bp;内含子数量共计28 187个,平均长度为118.94 bp;内含子合计长度为3.35 Mb,占基因组的7.65%。
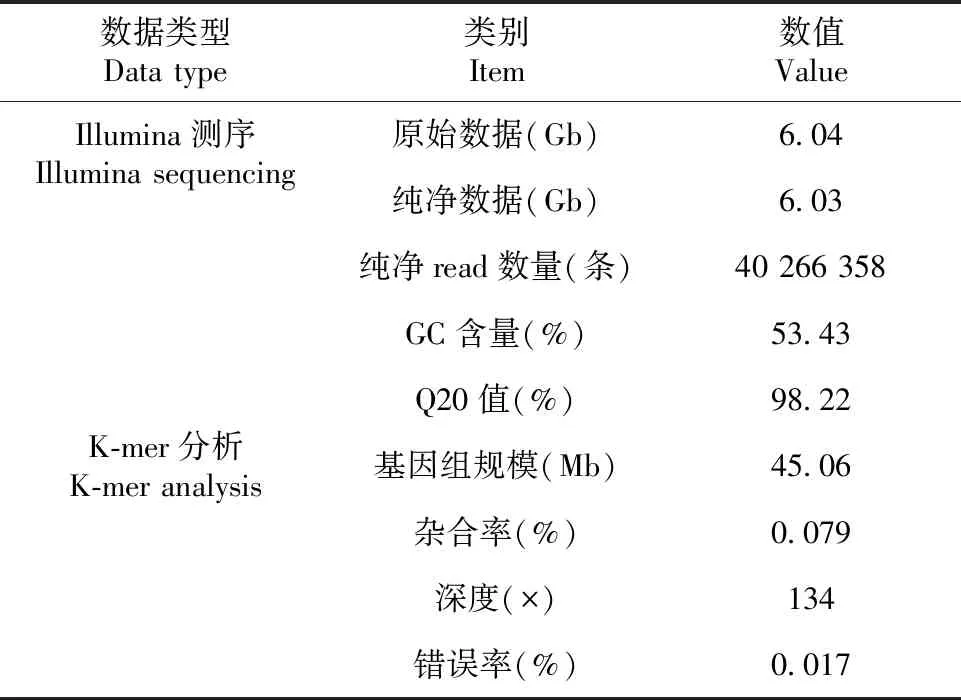
表1 N.dimidiatum H1基因组Illumina测序和K-mer分析

表2 组装的N.dimidiatum H1基因组BUSCO评估
2.4.2 非编码RNA 预测结果(表3)表明,N.dimidiatumH1基因组的非编码RNA含有核糖体RNA(rRNA)、小的调控RNA(sRNA)、核仁小RNA(snRNA)及转运RNA(tRNA)共计205个,总长度为98 500 bp,在基因组中占比0.2250%。其中,tRNA 108个,总长度10 166 bp,在基因组中占比0.0232%;rRNA 43个,总长度为79 409 bp,在基因组中占比0.1814%;snRNA 51个,总长度为8092 bp,在基因组中占比0.0185%;sRNA 3个,总长度为833 bp,在基因组中占比0.0019%。
2.4.3 转座子和重复序列 从表4可知,N.dimidiatumH1基因组的转座子总量为2158个,长度为697 214 bp,占基因组的1.59%。其中,逆转座子中以长末端重复(LTR)和长散在元件(LINE)数量较多,其中,LTR类的Gypsy类型数量841个,长度为483 597 bp,占基因组的1.10%;Copia类型数量137个,长度为13 031 bp,占基因组的0.03%;LINE类数量585个,长度为150 268 bp,占基因组的0.34%。DNA转座子数量492个,长度为42 843 bp,占基因组的0.10%。重复序列预测:N.dimidiatumH1基因组的重复序列总数量16 960个,长度为707 932 bp,占基因组的1.62%。其中,简单重复序列数量最多,为15 318个,长度为624 367 bp,占基因组的1.43%;低复杂度重复序列数量1614个,长度为80 898 bp,占基因组的0.18%。
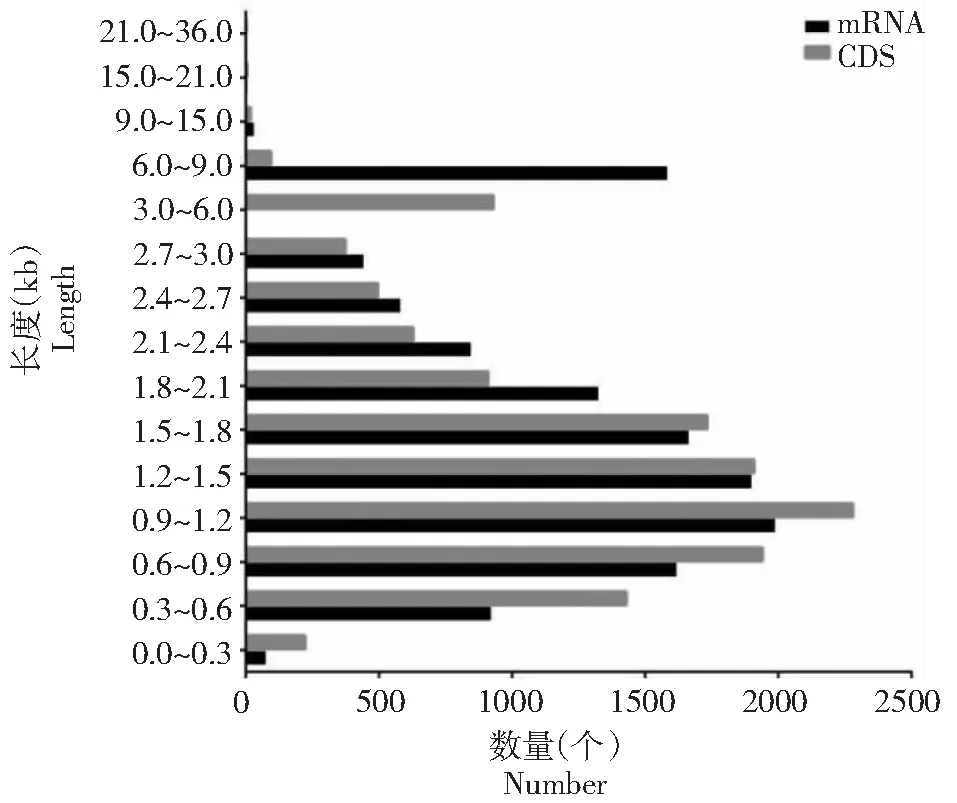
图1 N.dimidiatum H1基因组中mRNAs和CDS的长度分布
2.5 基因功能注释
从表5可知,预测获得的12 962个基因经Uniprot、Pfam、Refseq、Nr、Interproscan、GO、KEGG、Pathway和COG等9个数据库比对,共计12 703个(98.00%)基因被注释。其中,NR数据库注释的基因数最多,为12 643个,占总注释量的97.54%;其次分别为Interproscan数据库的10 443个(80.57%)和Pfam数据库的10 408个(80.30%);然后是Uniprot数据库的7956个(61.38%)和GO数据库的7882个(60.81%)。
2.5.1 NR注释 从图2看出,NR注释的物种分布中,以M.phaseolinaMS6的基因最多,占总注释量的58.36%;其余依次为N.parvumUCR-NP2(23.12%)、Diplodiacorticola(6.23%)和D.seriata
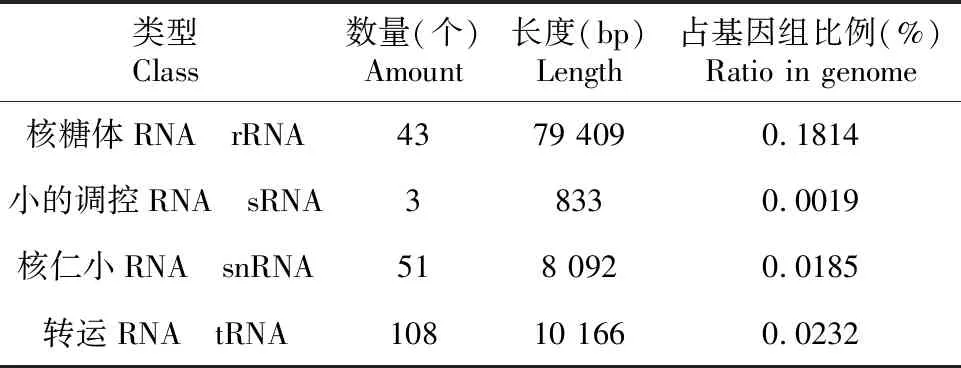
表3 N.dimidiatum H1基因组的非编码RNA预测
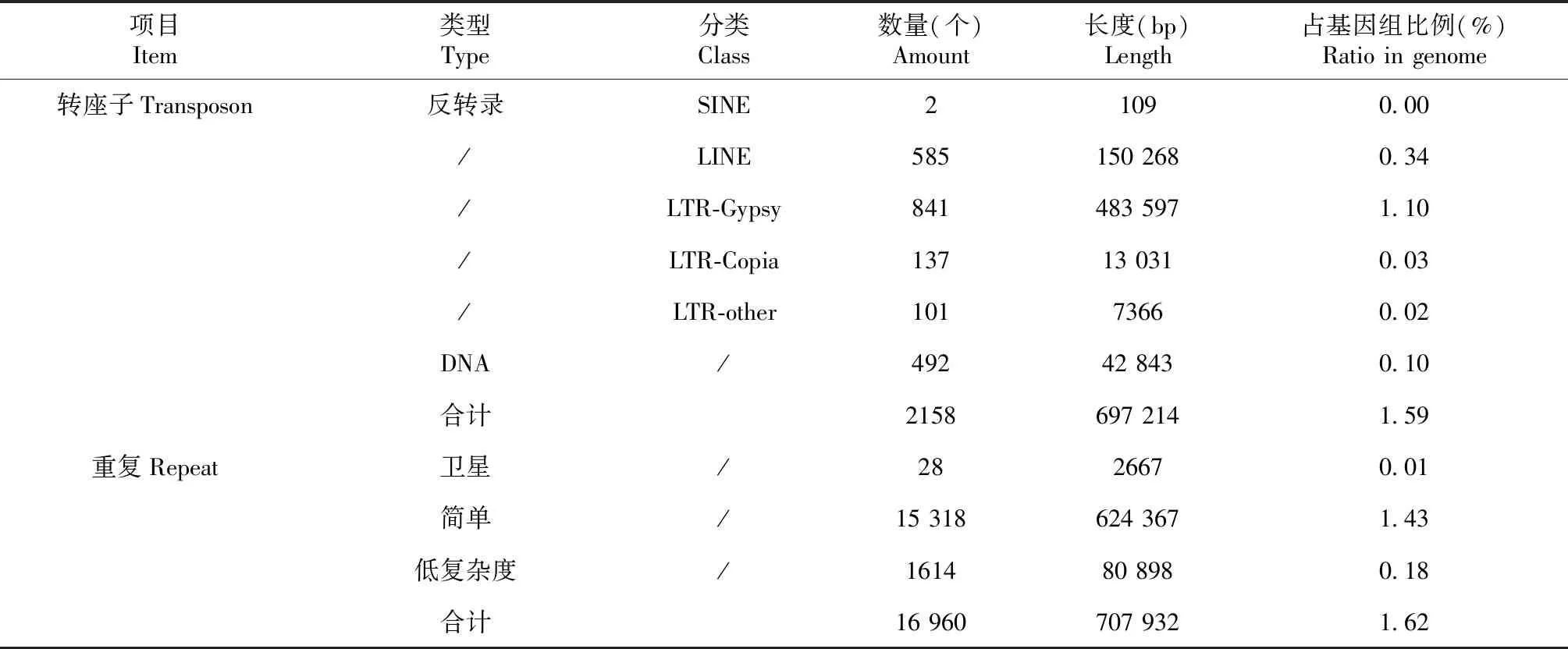
表4 N.dimidiatum H1基因组的转座子和重复序列预测

表5 N.dimidiatum H1基因组编码基因注释

A:物种分布;B:E值分布;a,0~1e-150;b,1e-150~1e-100;c,1e-100~1e-50;d,1e-50~1e-20;e,1e-20~1e-5;C:序列一致性分布。
(5.82%)。NR注释的序列一致率分布中,以80%~95%的基因数量最多,占51.88%;其余依次为60%~80%(27.26%)、40%~60%(9.77%)和95%~100%(8.76%)。NR注释的E值分布中,0~1e-150、1e-150~1e-100和1e-100~1e-50的基因数量占比分别为59.85%、15.85%和15.38%。
2.5.2 GO注释 统计GO注释(图3)表明,在生物学过程的70个二级分类中,生物合成过程(1851个)、细胞含氮化合物代谢过程(1330个)、生物过程(1172个)、催化过程(1085个)和小分子代谢过程(962个)是数量最多的5个二级分类,占比分别为23.48%、16.87%、14.87%、13.77%和12.21%。在细胞组分的35个二级分类里,细胞核(2050个)、细胞质(1671个)、细胞组分(1630个)、胞浆(1362个)和蛋白复合体(1357个)是数量最多的5个二级分类,占比分别为26.00%、21.20%、20.68%、17.28%和17.22%。在分子功能的41个二级分类里,离子结合(3030个)、分子功能(2054个)、 氧化还原活性(1472个)、跨膜转运活性(744个)、DNA结合(620个)是数量最多的5个二级分类,占比分别为38.44%、26.05%、18.68%、9.44%和7.87%。
2.5.3 KEGG注释 统计KEGG注释(图4)表明,31个二级分类中,碳水化合物代谢相关基因数量最多,为445个,占比为15.78%;其次分别为氨基酸代谢(417个)、翻译(365个)、全局及概要图(338个)、转运和分解(311个),占比分别为14.79%、12.94%、11.99%和11.03%;折叠、分类和降解(296个)、信号转导(253个)、脂类代谢(250个)、能量代谢(205个)及有害异物生物降解和代谢(202个),占比分别为10.50%、8.97%、8.87%、7.27%和7.16%。
3 讨 论
葡萄座腔菌科属子囊菌门(Ascomycota)座囊菌纲(Dothideomycetes)葡萄座腔菌目(Botryosphaeriales),是林木和园艺作物溃疡病的重要真菌性病原,寄主种类多样,地理分布广泛[28-30]。葡萄座腔菌科中一些危害园艺作物的属,如葡萄座腔菌属(Botryosphaeria)[24,31-32]、壳球孢属(Macrophomina)[23,33]、色二孢属(Diplodia)[34]、新壳梭孢属(Neofusicoccum)[35]和毛色二孢属(Lasiodiplodia)[36-37]均有若干种或株系已报道基因组序列,为了解病原菌的系统分类、遗传变异和致病机理奠定了基础。新节格孢属(Neoscytalidium)于2006年确立,目前仅包含N.dimidiatum、N.novaehollandiae和N.orchidacea-rum[38-39]。N.dimidiatum是新节格孢属的典型种[38],近年来在园艺作物普遍发生[13-15]。研究表明,N.dimidiatum引起人类皮肤病或鼻窦炎等[16-17],Santana等[39]从患者皮肤分离N.dimidiatumUM880株系开展基因组测序。目前尚未有分离自园艺作物的N.dimidiatum基因组报道,本研究从火龙果溃疡病中分离到病原新暗色柱节孢(N.dimidiatum) H1菌株并对其开展基因组测序和组装分析。结果表明,火龙果溃疡病病原N.dimidiatumH1基因组包含14个contig,N50长度为3.92 Mb,contig最长长度为5.63 Mb。与葡萄座腔菌科其他真菌测序策略和组装结果比较后发现,与采用Illumina测序策略(仅构建0.5 Kb文库)获得的B.dothideaCBS115476[28]、LW030101[31]和PG45[32]、DiplodiaseriataDS831[34]、N.parvumUCR-NP2[35]和LasiodiplodiatheobromaeLA-SOL3[37]基因组相比,H1基因组的contig长度大幅增加,且数量明显减少。与采用Illumina测序策略(构建0.5 Kb文库+5~6 Kb文库)获得的LasiodiplodiatheobromaeCSS-01s[36]和N.dimidiatumUM880[40]基因组相比,N50长度基本一致,而H1基因组的contig数量略少。与采用PacBio+Illumina或454+Illumina混合策略获得的B.dothideasdau11-99[24]和LW-Hubei(GenBank登录号:GCA_011064635.1)、M.phaseolina11-12[23]、Al-1[23]和MS6[33]基因组相比,N50长度也基本一致。进一步分析Illumina序列的比对率和覆盖率,结合真核和真菌直系同源基因的BUSCO评估,均表明组装的H1基因组完整性较高。因此,本研究获得的N.dimidiatumH1基因组与采用PacBio+Illumina或454+Illumina策略获得的基因组组装水平基本一致,contig长度较长,完整性较高,为开展后续研究奠定了良好基础。
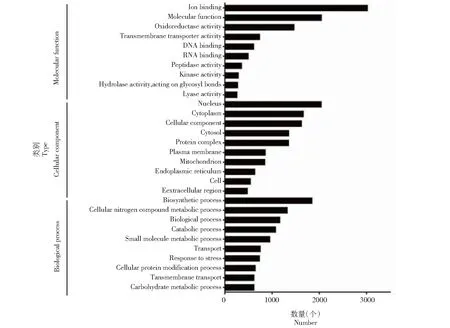
图3 N.dimidiatum H1基因组编码基因的GO分类
组装获得N.dimidiatumH1的基因组大小为43.78 Mb,与B.dothideaCBS115476[28]、L.theobromaeCSS-01s[36]和LA-SOL3[37]基本一致,但略高于分离自人体的N.dimidiatumUM880[40]。H1基因组含有的编码基因数量和基因密度均与L.theobromaeCSS-01s[36]和LA-SOL3[37]较一致,略高于N.dimidiatumUM880[40]。H1基因组含有108个tRNA,低于B.dothideaPG45[32]和N.dimidiatumUM880[40];HI基因组含有43个rRNA,介于N.dimidiatumUM880[40]和B.dothideasdau 11-99[24]之间。N.dimidiatumH1基因组中的重复序列和转座子占比均明显低于M.phaseolinaMS6[33],但转座子占比显著高于N.dimidiatumUM880[40]。在逆转座子类型中,N.dimidiatumH1基因组以LTR类Gypsy亚族和LINE类为主要类型,与N.dimidiatumUM880、M.phaseolinaMS6及其他真菌中主要逆转座子的类型均一致[33,40-41]。但N.dimidiatumH1基因组中的LTR类Gypsy亚族和LINE类的占比均明显高于N.dimidiatumUM880[40]。基于上述基因组结构上的初步比较表明,分离自火龙果的N.dimidiatumH1菌株与分离自人类皮肤的N.dimidiatumUM880的基因组存在一定差异。
N.dimidiatumH1的基因组中含有12 962个编码基因,共计98%的基因获得注释。NR注释的物种主要为葡萄座腔菌科的M.phaseolinaMS6和N.parvumUCR-NP2;NR注释的序列一致率主要分布在60%~95%;E值主要分布在0~1e-50,说明同源性较高,注释结果可靠。N.dimidiatumH1的基因组GO注释基因数量与N.dimidiatumUM880的基本一致,氧化还原酶活性、跨膜转运蛋白活性等基因数量基本一致,但离子结合相关基因数量远大于N.dimidiatumUM880[40]。KEGG注释表明,N.dimidiatumH1的基因组含有碳水化合物代谢、氨基酸代谢、脂类代谢、有害异物生物降解和代谢等基因,与N.dimidiatumUM880基本一致[40]。下一步需深入开展基因功能注释,如预测真菌致病相关的分泌蛋白及其包括的效应蛋白、碳水化合物酶类[42-44],为筛选致病基因和开展致病机理研究奠定基础。
4 结 论
利用Nanopore+Illumina混合测序策略对分离自火龙果溃疡病的N.dimidiatumH1开展基因组测序、组装和注释,获得N.dimidiatumH1的基因组大小为43.78 Mb,组装获得14个contig,N50长度为3.92 Mb,最长长度为5.63 Mb。N.dimidiatumH1的基因组含有12 962个编码基因,12 703个编码基因被注释,与同属于葡萄座腔菌科(Botryosphaeriaceae)的MacrophominaphaseolinaMS6和NeofusicoccumparvumUCR-NP2具有较高的同源性。该研究获得高质量N.dimidiatumH1的基因组序列,为了解N.dimidiatum的遗传变异、致病机理和制定防控策略奠定了理论基础。
猜你喜欢
发明与创新(2021年39期)2021-11-05
今日农业(2021年11期)2021-08-13
基层中医药(2021年2期)2021-07-23
今日农业(2019年14期)2019-01-04
现代园艺(2018年3期)2018-02-10
现代园艺(2018年14期)2018-01-18
小布老虎(2016年18期)2016-12-01
小主人报(2015年23期)2015-02-28
遗传(2014年3期)2014-02-28
世界科学(2014年8期)2014-02-28
