
分散型钼基催化剂在煤显微组分与催化裂化油浆共加氢反应中的应用
2023-09-06 12:48涂椿滟秦鹏伟刘昌伟
石油炼制与化工 2023年9期
涂椿滟,秦鹏伟,刘昌伟,黄 伟
(太原理工大学化学工程与技术学院,省部共建煤基能源清洁高效利用国家重点实验室,太原 030024)
煤与劣质重油(如催化裂化油浆)加氢共处理技术能够同时实现煤炭清洁高效转化和重油加工利用,对降低我国石油对外依存度、保障国家能源安全具有重要意义[1-2]。煤与重油共加氢反应原料通常采用芳香度较高的重油与煤化程度较低的褐煤或次烟煤[3-5]。将不同等级煤作为原料和重油进行加氢共炼,最终获得的煤转化率有明显差异[5],适宜于煤/油共炼的煤种选择范围有限。对于不同产地的煤,因化学组成、矿物质特征、显微组分差别较大,导致其具有不同的反应性能。煤中有机显微组分包括壳质组、镜质组和惰质组,一般认为,低阶煤中镜质组和壳质组比热稳定性较高的惰质组容易转化,但有文献报道在Fe基催化剂作用下,经原煤分离得到的纯镜质组(质量分数大于95%)与蒽油加氢共炼时的煤转化率和油收率均低于原煤与蒽油加氢共炼时的煤转化率和油收率[6-7]。目前关于煤显微组分液化反应性的认识尚存在矛盾,有必要开展进一步的研究。
催化剂作为煤与重油加氢共处理的关键因素,在降低悬浮床工艺操作苛刻度、提升煤转化深度、提高液体油收率和品质等方面发挥着重要作用。传统加氢催化剂中的活性金属组分包括Mo,Ni,Fe,Co,W等[8-12]。Fe系催化剂虽然价格低廉,但其催化活性相对较低,这导致煤直接液化的油收率偏低、固体残渣量高[13-14]。Mo系催化剂的活性组分为具有S-Mo-S层状结构的MoS2,活化H2的能力较强,加氢活性高。已有研究表明在渣油悬浮床加氢裂化中,分散型Mo催化剂表现出了良好的催化性能,催化剂前躯物分散在反应油中原位生成MoS2,其抑制焦炭生成能力和液体产率均优于Ni,Fe,Co催化剂[15-18]。因此,将分散型Mo基催化剂应用于煤与重油悬浮床加氢共处理中具有很大的潜力。
本研究首先制备四硫代钼酸铵(MS),然后利用其与正辛基三甲基氯化铵之间的烷基取代反应合成正辛基三甲基硫代钼酸铵(OT-MS)。通过X射线衍射(XRD)、电感耦合等离子体发射光谱(ICP-OES)、傅里叶变换红外光谱(FTIR)、N2物理吸附和透射电子显微镜(TEM)对两种分散型Mo催化剂进行结构分析。以凉水井原煤(LSJR)和催化裂化(FCC)油浆为反应原料,对比研究MS和OT-MS在煤与重油共加氢中的催化活性。以OT-MS为Mo催化剂前躯体,选取LSJR及其惰质组富集物(LSJ-I)和镜质组富集物(LSJ-V)3种煤样为原料,分别与FCC油浆进行共加氢反应,研究煤中显微组分组成对共加氢产物分布及液体产物性质的影响。
1 实 验
1.1 原 料
四水合钼酸铵((NH4)6Mo7O24·4H2O),分析纯,上海阿拉丁生化科技有限公司产品;氨水、正辛基三甲基氯化铵([C8H17(CH3)3]NCl,OTAC),均为分析纯,上海麦克林生化科技有限公司产品;硫化铵水溶液、甲苯、正庚烷、乙醇,均为分析纯,国药集团化学试剂有限公司产品。
选取LSJR作为原料煤样,对其进行煤显微组分富集和宏量分离处理,得到纯度较高的LSJ-I和LSJ-V,具体分离方法见文献[19]。以上3种煤样均用作煤与重油共炼原料,其工业分析、元素分析和煤岩分析结果见表1。试验用油为陕西延长石油(集团)有限公司提供的FCC油浆,其具体性质见表2。
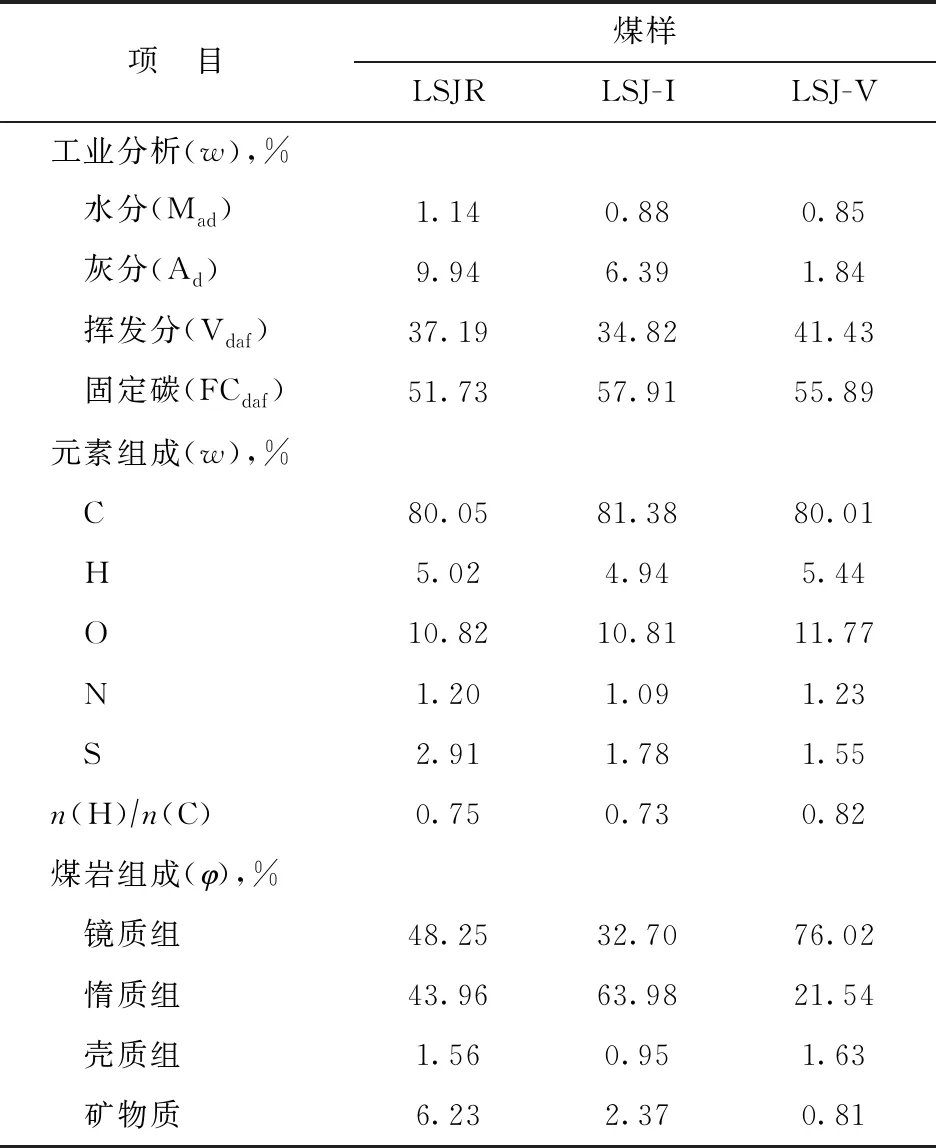
表1 实验用煤的性质
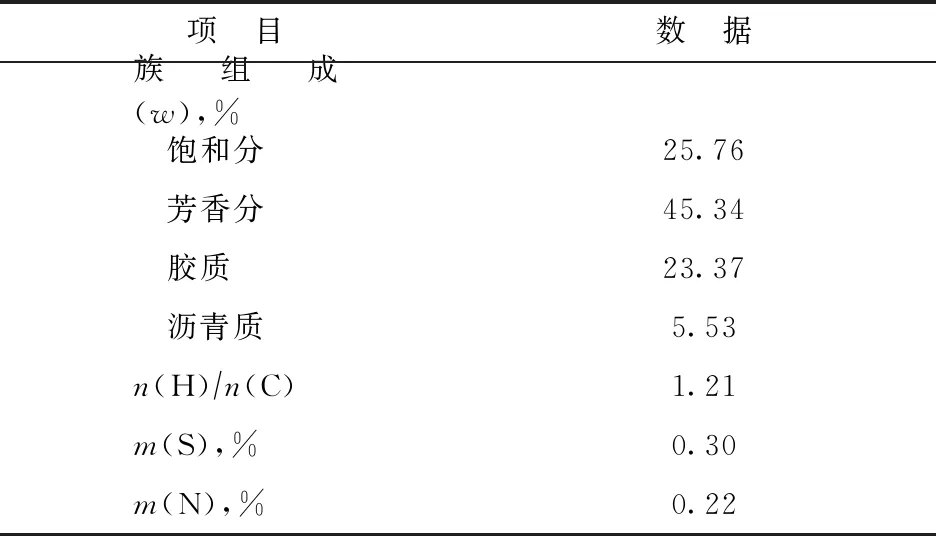
表2 FCC油浆的性质
1.2 催化剂制备
将一定量四水合钼酸铵溶解于氨水溶液中,以n(S)/n(Mo)为4∶1的比例加入硫化铵水溶液。将该混合物于70 ℃下搅拌1 h,然后在冰水浴中冷却结晶。经冰水和冰乙醇洗涤、60 ℃真空干燥后得到红棕色晶体MS。
取上述合成的MS样品,加入到预先配制的OTAC水溶液中,其中n(OTAC)/n(Mo)为2.2∶1。将混合物在40 ℃下搅拌1 h,然后在冰水浴中冷却,经洗涤、真空干燥得到OT-MS。
1.3 催化剂表征
采用D/max 2500型X射线衍射仪(日本Rigaku公司产品)对样品进行物相分析,Cu钯Kα射线,扫描速率为4(°)/min,采集范围为5°~70°。样品中Mo含量在Agilent 730 ICP-OES光谱仪(美国Agilent公司产品)上进行测定。红外光谱表征在Tensor 27型红外光谱仪(德国Bruker公司产品)上进行,扫描范围为400~4 000 cm-1。利用Quadrasorb SI型物理吸附分析仪(美国Quantachrome公司产品)测定样品的比表面积和孔径分布,N2吸附-脱附试验在液氮温度下进行。利用JEM-2100F型透射电子显微镜(TEM,日本JEOL公司产品)观察样品中MoS2晶粒的形貌,随机拍摄15张照片,至少统计500个MoS2晶粒。
1.4 煤与重油加氢共炼反应
煤与重油加氢共炼反应在100 mL高压釜中进行。称取6.0 g原料煤(干基煤)、18.0 g FCC油浆装入反应釜中,催化剂添加量(以Mo金属计)为反应原料总质量的0.1%。将釜内空气置换干净后,通入H2充压至8.0 MPa。开启搅拌和加热,搅拌速率为800 r/min,在420 ℃下反应60 min后结束。待釜内温度降至室温后,收集釜内所有产物。使用甲苯对部分液-固产物进行索氏抽提,甲苯不溶物为固体残渣,对残渣进行烧灰分析。按照下式计算气体收率yG、液体收率yL、固体收率yS以及干基无灰煤转化率x。
(1)
(2)
yG=100%-yL-yS
(3)
(4)
式中:m0为原料干基煤和FCC油浆的总质量,g;m1为液-固产物总质量,g;ws为液-固产物中所含固体残渣的质量分数,%;mdaf为干基无灰煤质量,g;wash为固体残渣中灰分的质量分数,%。
2 结果与讨论
2.1 催化剂的表征分析
图1为合成的两种Mo催化剂前躯体MS和OT-MS的XRD图谱。由图1可知,MS样品在2θ为14.5°,17.2°,18.5°,23.5°,25.2°,31.6°等处出现明显的归属于(NH4)2MoS4物种的衍射峰,峰形尖锐,表明合成的MS结晶性较好。OT-MS样品中并未观察到明显的特征衍射峰,呈现无定形状态。表3列出了两种催化剂前躯体中Mo的理论含量和ICP-OES分析结果,其中MS和OT-MS的理论Mo含量分别根据其化学式(NH4)2MoS4和[C8H17N(CH3)3]2MoS4进行计算。由表3可见,两种催化剂前躯体中Mo含量的实测值均与理论值符合较好。结合XRD分析结果可知,合成的催化剂前躯体纯度较高。将上述两种Mo催化剂前躯体样品在氢气气氛中于420 ℃温度下处理1 h,MS和OT-MS的热分解产物分别记作MS-c、OT-MS-c。从图1可以看出,MS-c样品在2θ分别为14.1°,33.5°,59.1°处的衍射峰对应MoS2物种的(002),(100),(110)晶面[20],峰形较宽,而OT-MS-c样品仅在2θ为33.5°和59.1°处呈现微弱的MoS2特征衍射峰,这表明两种催化剂前躯体受热分解后均转变成MoS2微晶,且OT-MS-c中MoS2晶粒粒径更小。
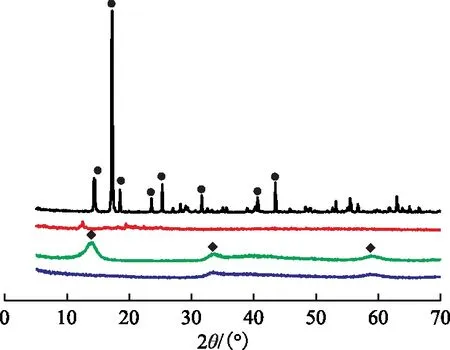
图1 催化剂前躯体MS、OT-MS及其分解产物MS-c、OT-MS-c的XRD图谱
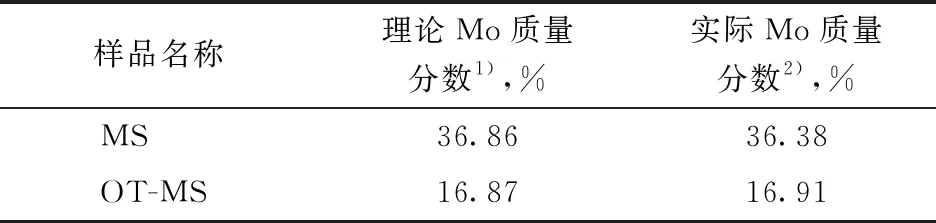
表3 催化剂前躯体MS和OT-MS中Mo的理论含量和实际含量
对两种Mo基催化剂前躯体进行FTIR表征,结果如图2(a)所示。由图2(a)可以看出,不同于MS样品,OT-MS在波数为2 922 cm-1和2 850 cm-1处有明显的吸收峰,分别代表—CH2的不对称和对称伸缩振动,还在波数为1 463 cm-1处有—CH2的弯曲振动吸收,进一步说明OT-MS中存在烷基链结构。MS的Mo—S键伸缩振动出现在波数为480 cm-1附近,峰形尖锐,而OT-MS则在较低波数467 cm-1处呈现Mo—S键的伸缩振动吸收,且强度较弱,这暗示了将烷基基团引入MS后,因烷基相较于氢原子的供电子效应更强,使得Mo—S键强度变弱[21]。图2(b)为两种Mo基催化剂前躯体分散在二氯甲烷中的照片。由图2(b)可见,MS不溶于二氯甲烷且沉积在底部,而OT-MS则形成了澄清的棕红色溶液,说明催化剂前躯体OT-MS具有良好的油溶性。

图2 催化剂前躯体MS、OT-MS的FTIR谱图及其分散在二氯甲烷中的照片
利用N2物理吸附表征对比了不同前躯体制得的MoS2的孔结构,N2吸附-脱附曲线和孔径分布如图3所示,结构参数见表4。由图3可以看出,OT-MS-c呈现为Ⅳ型等温线并含H2型滞后环,具有丰富的介孔结构,孔径主要分布在3~6 nm范围内。由表4可知,与MS-c相比,OT-MS-c样品具有更大的比表面积和孔体积,分别达到469 m2/g和0.53 cm3/g,表明在MS中引入烷基链有助于形成具有高比表面积和孔体积的MoS2。

图3 MoS2样品的N2吸附-脱附曲线和孔径分布

表4 MoS2样品的结构参数
TEM表征被用于观察MoS2的微观形貌。图4为样品MS-c和OT-MS-c的TEM照片,图中呈现的黑色条纹为MoS2片层,样品均由具有纳米片层结构的MoS2堆叠而成。现有研究认为[16,22-24],MoS2的催化活性来源于MoS2片晶的边缘部分,基于“Rim-Edge”模型,MoS2晶粒的尺寸越小,堆叠层数越少,则暴露的Rim活性位越多,加氢活性越高。图5为MoS2晶粒的片晶长度和堆叠层数分布情况的统计结果。由图5可以看出:样品MS-c中MoS2片晶的长度主要分布在4~7 nm,片晶堆叠程度较高且层数分布较宽,70.1%的MoS2晶粒为3~5层;样品OT-MS-c中MoS2片晶的长度集中分布在3~6 nm,堆叠程度较低,超过90%的MoS2晶粒仅有1~3层。由此可知,在MS中引入烷基链会使最终形成的MoS2的活性相形貌结构呈现显著差异。与MS-c相比,OT-MS-c中MoS2片晶长度短、堆叠层数少且分布窄,活性金属的分散性好。结合前文XRD表征结果可知,催化剂前躯体OT-MS在受热分解过程中形成了大量的小尺寸MoS2晶粒,能够暴露出更多的Rim活性位,有助于提高催化加氢反应活性。

图4 MoS2样品的TEM照片
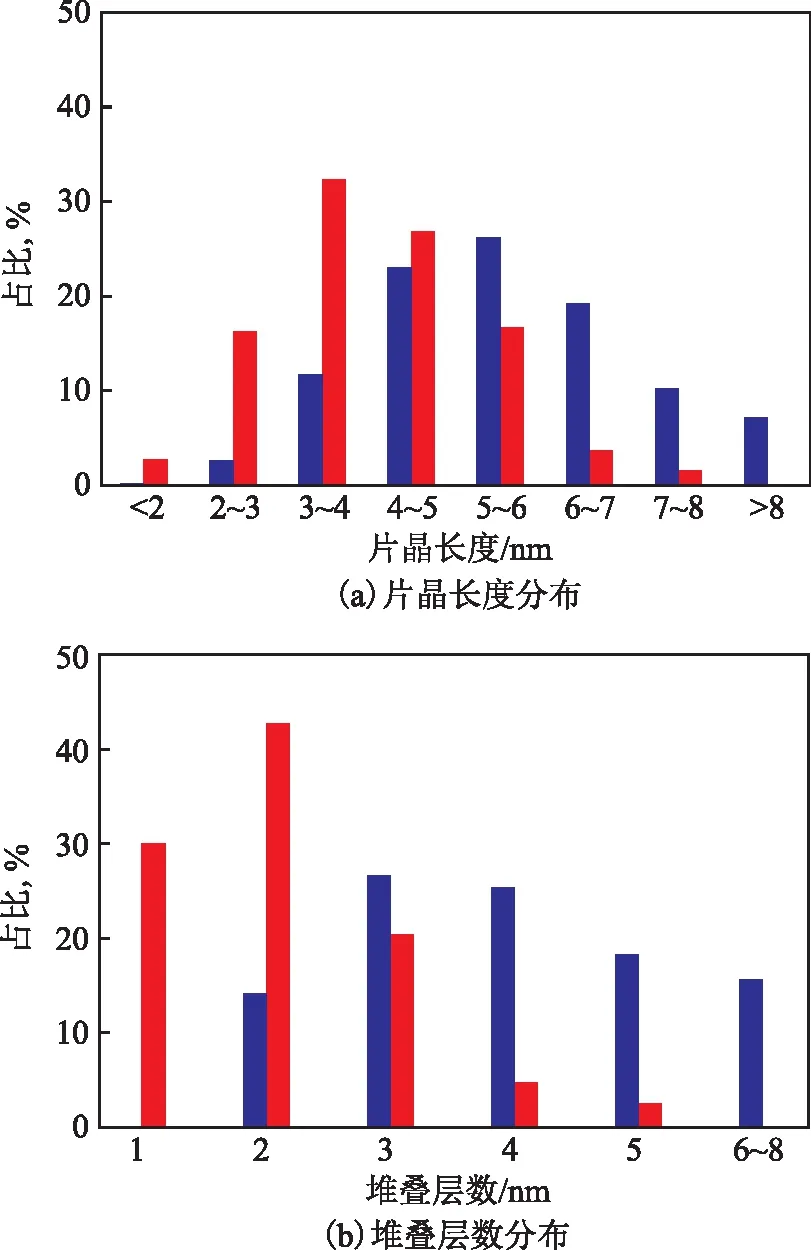
图5 MoS2晶粒的片晶长度和堆叠层数分布
2.2 煤显微组分与重油共加氢反应性能
以凉水井原煤为原料煤、FCC油浆为原料油,研究分散型Mo基催化剂在煤与重油共加氢反应中的催化性能。不同催化剂作用下的共加氢反应产物分布、干基无灰煤转化率以及反应氢耗见表5。从表5可以看出,反应体系中不添加催化剂时,煤转化率仅为42.9%,气体和固体产物收率分别为7.0%和15.4%。而添加少量Mo催化剂后,煤转化率和共加氢液体产物收率均明显升高,气体、固体收率下降,同时反应氢耗增加。这可能是由于催化剂对氢气的活化作用导致活性氢原子的大量生成,促进了热裂化产生的自由基碎片与活性氢原子的结合,从而有效抑制自由基碎片缩聚生成焦炭,提高了煤的液化程度。与MS相比,OT-MS在原煤与重油共加氢反应中表现出更高的催化活性,干基无灰煤转化率提升至77.1%,共加氢液体产物收率达到89.0%,固体残渣收率降低至8.3%。

表5 煤显微组分与FCC油浆加氢共处理的反应结果
结合前文中催化剂表征结果可知,相较于MS,具有油溶性的OT-MS更易于在煤/油反应体系中均匀分散,使得催化剂与反应原料的接触几率增大;由OT-MS形成的MoS2具有更大的比表面积,有利于反应物的扩散,且活性金属分散性好,大量的小尺寸MoS2晶粒对氢气的活化能力强,能够为共加氢反应体系提供更多的活性氢自由基,抑制结焦反应,从而提升了原料中重质组分的转化程度以及共加氢反应的液体收率。
分别以LSJ-I、LSJ-V作为共炼原料煤,研究OT-MS催化作用下煤显微组分与FCC油浆共加氢的反应特性,结果如表5所示。由表5可见,采用LSJ-I时煤转化率为68.0%,较原煤LSJR反应时降低了9.1%,且气体、固体收率有所增加。相较于原煤和FCC油浆共处理,采用LSJ-V进行反应时煤转化率由77.1%提升至90.3%,固体残渣收率由8.3%降低至3.4%,且液体收率和反应氢耗明显增加。从表1可以看出,LSJ-I,LSJR,LSJ-V的镜质组质量分数分别为32.70%,48.25%,76.02%,挥发分及H/C原子比的大小顺序均为LSJ-I 为了分析煤显微组分对煤/油共加氢液体产物性质的影响,分别测定了使用不同显微组分富集煤样时所得的液体产物的族组成以及H/C原子比,结果如图6所示。由图6可以看出,与不添加催化剂相比,向原煤LSJR与重油共转化的反应体系中添加OT-MS后,液体产物油中胶质、沥青质含量大幅度减少,饱和分、芳香分含量明显增加,且H/C原子比增大。这意味OT-MS的存在促进了胶质、沥青质的裂解,以及芳烃的加氢饱和及裂解开环,使轻质油组分增加。由图6可见,在OT-MS催化作用下,使用3种不同煤样LSJ-I、LSJR和LSJ-V作为反应原料煤时,共加氢液体产物的族组成以及H/C原子比存在明显差别。随着原料煤中镜质组含量的提高,共加氢液体产物中饱和分含量逐渐上升,胶质、沥青质含量减少,H/C原子比则呈现逐渐增大的趋势。采用LSJ-V与重油共转化体系时所得共加氢液体产物中沥青质含量最低,H/C原子比最高,油品质较好。 图6 OT-MS催化不同煤显微组分与FCC油浆共加氢反应液体产物的族组成和H/C原子比 (1)通过不同前躯体(MS、OT-MS)形成的MoS2的孔结构和活性相形貌有明显差异。在MS中引入烷基链有助于形成高比表面积的MoS2。与MS相比,由OT-MS形成的MoS2片晶长度短、堆叠层数少且分布窄,活性金属的分散性好。 (2)对于凉水井原煤和FCC油浆的共加氢反应,添加分散型Mo催化剂使干基无灰煤转化率和共加氢液体产物收率均得到了明显的提升,气体、固体残渣收率减少。与MS相比,OT-MS表现出更高的催化活性,共加氢液体产物和固体残渣收率分别为89.0%和8.3%,干基无灰煤转化率为77.1%。在OT-MS催化作用下,煤显微结构对煤与重油共加氢产物分布和液体产物油的品质有显著影响。 (3)采用不同显微组分富集煤样进行共加氢反应,干基无灰煤转化率和共加氢液体产物收率从低到高顺序均为:惰质组富集物LSJ-I<凉水井原煤LSJR<镜质组富集物LSJ-V。随着原料煤中镜质组含量的提高,液体产物中饱和分含量上升,胶质、沥青质含量下降,H/C原子比增大,轻质化效果更明显。
3 结 论
猜你喜欢
能源化工(2022年3期)2023-01-15
石油沥青(2022年4期)2022-09-03
能源化工(2021年6期)2021-12-30
宁夏大学学报(自然科学版)(2021年2期)2021-07-28
能源工程(2021年1期)2021-04-13
船舶标准化工程师(2020年1期)2020-06-12
西安科技大学学报(社会科学版)(2019年6期)2019-09-10
河北工程大学学报(自然科学版)(2015年4期)2016-01-26
化工进展(2015年11期)2015-07-24
滁州学院学报(2014年2期)2014-07-12
