
重质船用燃料油的沥青质组分在柴油中的相行为模拟研究
2023-09-06 12:34师黎明
石油炼制与化工 2023年9期
师黎明,杨 鹤,任 强,李 妍,龙 军
(中石化石油化工科学研究院有限公司,北京 100083)
重质船用燃料油(重质船燃)是一种用于大型远洋船舶发动机的重质燃料,具有黏度大、热值高等特性。重质船燃主要采用重质油与轻质油调合[1-2],然而受国际海事组织(IMO)限硫协议及其他诸多因素影响[3-4],重质船燃的调合原料来源变得更加广泛,从石油炼制产物到深加工原料[5],甚至包括煤焦油、轮胎油等[6],然而,不同来源调合原料的组成存在明显差异,常常会影响原料间的相容性以及各组分的分散性,从而加剧重质船燃中沥青质的聚集与沉积,导致调合的重质船燃产品稳定性不佳[7-9]。
鉴于此,国内外学者尝试使用不同的原料进行重质船燃调合试验,优选出不同组分间的相容性较好、产品相对稳定的重质船燃调合方案。薛倩等[10]研究发现,在由煤焦油组分与沥青调合的重质船燃中掺入C9或C10低凝点高芳香性组分后,产品中的不稳定组分明显减少;Kass等[11]评估发现以生物质油与高黏度重质燃料(HFO)混合制备船用燃料油是可行的;Vráblík等[12]向由减压渣油(VR)与常压瓦斯油(AGO)调合的船用燃料油中加入轻循环油(LCO)或萘与四氢萘的混合物后,其加速老化沉积数值(TSA)均降低;Kondrasheva等[13]使用三角形相图发现由VR、超低硫柴油(ULSD)和LCO可以调合得到稳定性较好的船用燃料产品。
虽然通过调合试验探索可以获得稳定性较好的重质船燃,但以上结果多基于经验和实际操作获得,对其他原料不具有普遍的适用性,对于新的调合原料仍需进行复杂的试验探索。因此,从微观结构和分子水平认知重质船燃调合组分间作用的规律性,对于重质船燃产品的调合生产具有重要的指导意义。然而,由于重质船燃的组成极其复杂,目前还无法通过试验方法获得其真实的组成和组分结构。近年来,人工智能模拟方法被广泛用于重质油微观结构认知及其分子间相互作用的研究[14],如张文等[15]通过分子动力学(MD)模拟发现不同结构VR分子间的相互作用不同,从而导致其微观结构分布不均匀;关冬等[16]基于耗散粒子动力学(DPD)方法,对重质油体系沥青质微观聚集态进行模拟,将沥青质聚集率与胶体不稳定指数(CII)进行关联,提出了改进的重质油稳定性判据;Park等[17]采用MD方法对溶剂脱沥青(SDA)过程进行了模拟,发现SDA过程中尺寸较大的聚集体一般是由多个小聚集体聚集形成;Ramírez等[18]对PA3型结构沥青质分子在不同溶剂中的溶解行为进行了模拟研究,发现其分子中的杂原子能增加分子的极性和平面性,改善分子的聚集倾向。上述研究表明,借助分子模拟工具,可以深入探究重质船燃的微观组成结构及分子间相互作用情况,从而提升对重质船燃调合原料匹配规律的认知。
基于此,本研究采用MD等模拟方法,对重质船燃组分沥青质的分子结构、柴油调合原料的分子结构、不同分子间相互作用及组分间的相容性进行模拟计算,以提升对沥青质分子在柴油中相行为的认知和理解。
1 模拟方法
1.1 重质船燃分子模型
重质船燃分子模型的构建是基于典型调合原料的烃类组成特点进行的。其中,沥青质分子为塔河原油中沥青质的平均分子结构[19],其平均分子式为C47H41NOS2,相对分子质量为699;柴油原料主要有LCO和加氢精制柴油(简称加氢柴油)。研究表明[20]:LCO中的饱和烃组分主要为链烷烃,而其芳香烃组分以多环芳烃为主,因此选择与柴油主要组分特征相近的正十四烷、烷基萘及菲作为柴油模型分子;加氢柴油是由LCO选择性加氢得到,因而其饱和烃组分含有较多的环烷烃,其芳香烃组分则主要是单环芳烃,因此选择烷基十氢萘、烷基苯作为加氢柴油模型分子。重质船燃的各种模型分子结构如图1所示。其中灰、白、红、黄、蓝色球分别代表碳、氢、氧、硫、氮原子。
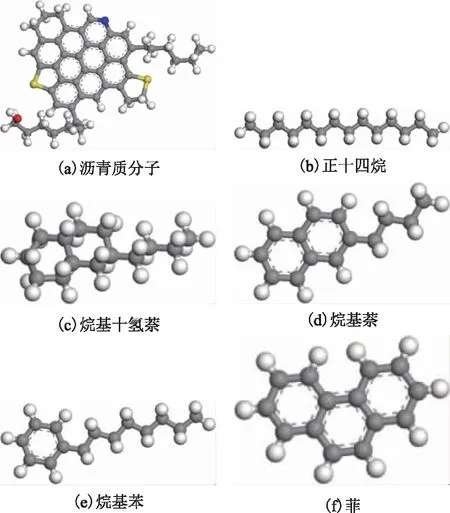
图1 重质船燃组分模型分子
1.2 量子化学计算方法
使用分子模拟软件Materials Studio 2017(简称MS)中的DMol3量子化学模块,优化模型分子的几何结构,并计算分子结构的静电势分布特性。分子结构优化过程中选择基于广义梯度近似(GGA)的PBE泛函,在大数值基组DNP(双数值轨道基组与p轨道极化函数)水平上进行全电子计算,自洽场(SCF)的迭代收敛阈值设置为1×10-5Ha,收敛精度为:能量2×10-5Ha,受力0.004 Ha/nm,位移5×10-4nm。对优化结果进行Potentials选项分析,得到分子结构的静电势分布。
1.3 蒙特卡洛抽样计算方法
使用MS中的Blends模块进行分子结合能的模拟计算:如图2所示,将两种不同结构分子(记作pack和seed)的质心进行重叠,之后移动pack分子,当二者的范德华表面不再重叠时,计算该空间构象下二者的结合能;再改变pack分子在空间的取向以及移动方向,重复上述计算过程,最终得到两种分子在空间里不同二聚体形貌的集合,对集合进行平均得到分子间的平均结合能(Ebs)。计算过程中,集合样本数取10 000,迭代次数选择20,计算环境温度选择373.15 K,为重质船燃的调合温度。
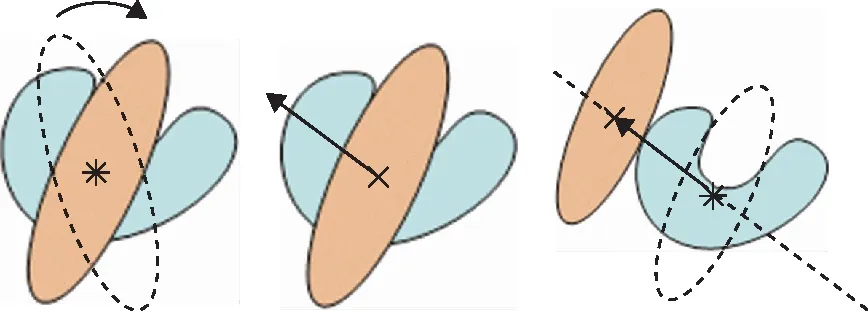
图2 Pack分子(黄色)与Seed分子(蓝色)的空间构象
1.4 分子动力学模拟方法
使用MS中的Amorphous Cell建立初始密度为0.4 g/L的无定形晶胞,并对晶胞进行结构优化与退火,选择退火过程中能量最低的构象。对低能量构象使用MS中Focite模块Dynamics任务项进行分子动力学模拟,先进行1 000 ps的NPT系综模拟以使体系达到合理的密度,再进行2 000 ps的NVT系综模拟使体系中的分子进行充分相互作用。当体系能量及其分项出现小范围波动时,表示体系已达到平衡。过程中力场选择COMPASSⅡ,计算精度为fine,静电作用和范德华作用的非键截断均采用Group based,温度与压力分别设置为373.15 K及标准大气压,且温度与压力的控制函数分别选择Nose与Berendsen。对充分平衡后的动力学模拟结果使用Cohesive Energy Density(CED)任务项进行溶解度参数的计算,计算式见式(1)[21]。
(1)
式中:Ecoh为沥青质内聚能,指将沥青质体系中的所有分子移动到无穷远处所需要的能量,kJ/mol;V为摩尔体积,cm3/mol;d为内聚能密度,J/cm3;δ为溶解度参数,(J·cm-3)0.5;ΔHv为沥青质分子气化热,kJ/mol;RT表示沥青质分子聚集体由固体或液体转化为气体时所需要的膨胀功,kJ/mol。
使用Energy任务项进行不同组分间平均作用能的计算,计算式见式(2):
Ea-b=Etotal-Ea-Eb
(2)
式中:Ea-b为a、b组分间的平均作用能,kJ/mol;Etotal为a、b组分共存时整个体系的总能量,kJ/mol;Ea与Eb则分别为a、b组分单独存在时体系的能量,kJ/mol。
对部分动力学模拟结果会进行径向分布函数(RDF)分析。RDF的意义是反映从一个任意指定的“中心”粒子出发,到半径为r的球壳层位置上出现其他粒子的概率[22],如图3所示。

图3 径向分布函数图示
本研究中以某一粒子为观测中心,以0.1 nm为球半径差(Δr)向外画出一层层的同心球壳,然后统计每一壳层里的粒子数密度与平均数密度的比值,即为径向分布函数,用g(r)表示,如式(3)。
(3)
式中:n(r)表示总粒子数;r为距中心原子半径,nm;V为体积,nm3;ρ0为理想晶体粒子数密度(ρ0=n(r)/V),δr为球壳层厚度,nm。RDF曲线越高,表明中心粒子周围越倾向于出现某种粒子。
2 结果与讨论
2.1 模型分子的静电势分布
对不同的重质船燃模型分子进行静电势分布模拟计算,过静电势分布推测分子的各种性质以及分子间的相互作用。各种模型分子的静电势模拟计算结果如图4所示。
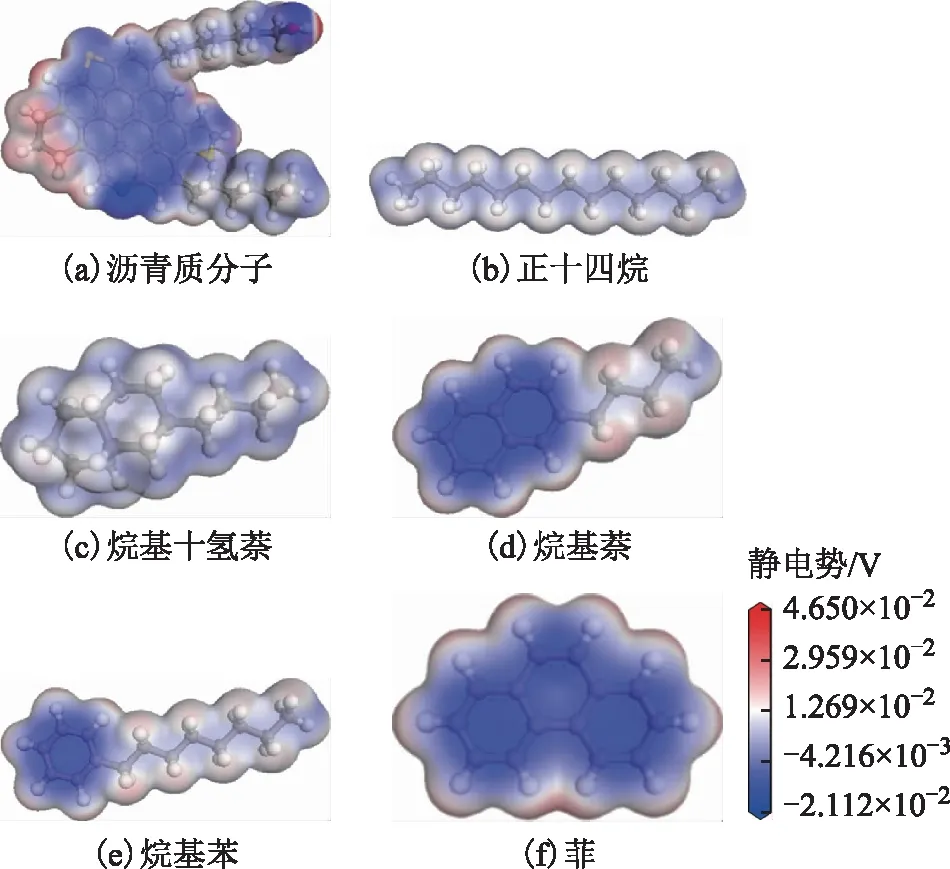
图4 模型分子的静电势分布
计算结果显示,不同来源的重质船燃模型分子,因其结构差异所呈现的静电势分布各不相同。沥青质分子因具有大芳环结构和多杂原子的结构特征,使得π电子云主要集中在芳环上,沥青质分子间因π电子云重叠而产生较强的π-π相互作用,在空间上会呈现出面对面式的堆叠形貌;而其芳环周围的H原子则带部分正电荷,可以与π电子云形成π-H作用使得分子间呈T型堆叠;沥青质分子杂原子芳环结构中的N原子以及芳环侧链上的羟基O原子带有负电荷,与羟基O原子相连的H原子带部分正电荷,使得沥青质分子间易形成O—H、N—H等类型的氢键作用。较小芳香烃模型分子的芳环结构中也因p轨道的肩并肩形成了大π键,π电子云在该结构中富集,且芳环数越多富集区域越大。芳香烃分子结构中的长烷基侧链以及饱和烃结构分子电荷分布较为均匀,分子间主要是范德华相互作用。对比不同结构分子间的静电势分布可以看到,在结构上芳香烃分子与沥青质分子较为相似,分子间容易形成π-π相互作用,而饱和烃结构分子与沥青质分子存在明显的结构差异,分子间可能主要形成范德华相互作用。
2.2 模型分子间的结合能
通过结合能的模拟计算可量化比较不同结构模型分子间的相互作用强弱,因此计算了沥青质分子自身及其与其他不同结构模型分子间的结合能,结果如图5所示。由图5可知,沥青质分子间具有很强的相互作用,最左侧黑色柱状图数值较高,在373.15 K下的分子间结合能为118.18 kJ/mol,因而其表现出很强的自聚倾向。不同结构的模型分子与沥青质分子的结合能存在明显的差异,其从高到低的顺序为菲>烷基萘>烷基苯>烷基十氢萘>正十四烷。由此可见:柴油原料中多芳环结构的菲、烷基萘分子与沥青质分子的结合能较高,表明芳环间的π-π相互作用能较高,这是因为π电子云重叠面积越大,π电子的离域范围越大,致使体系能量降低、稳定性提高;而含长烷基侧链的烷基苯分子及饱和烃分子与沥青质分子的结合能较低,可能的原因是长烷基侧链结构和饱和烃长链不利于接近沥青质分子,分子间形成的范德华作用相对较弱。
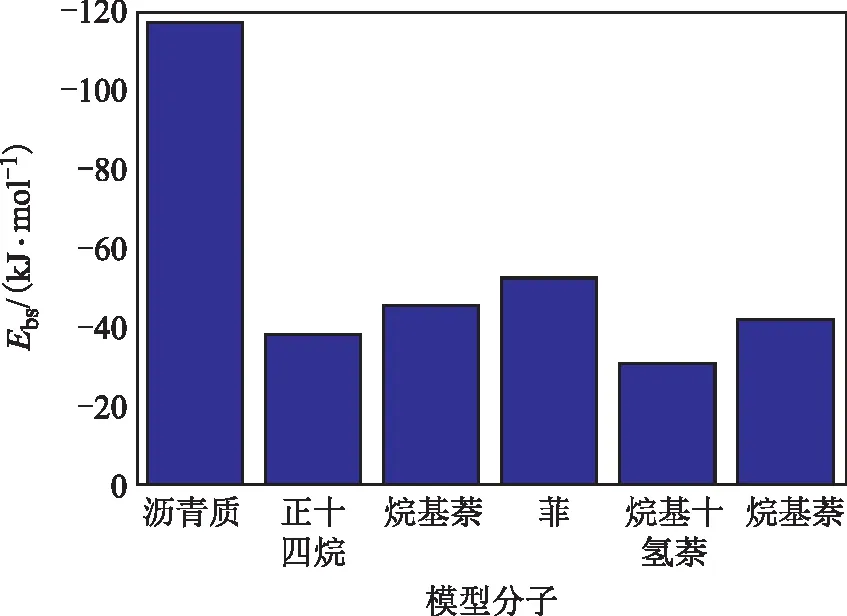
图5 重质船燃模型分子间的结合能
2.3 模型分子的溶解度参数
虽然分子结合能能够对单个分子间的相互作用强弱进行量化描述,但不能反应不同分子间的相行为。溶解度参数(δ)是衡量分子间相容性的重要参数,是对“相似相溶”原理的量化表征,因而通过溶解度参数的模拟计算可以预测重质船燃中不同结构分子间的相容性,能在一定程度上反映分子在溶液中的相行为。分子的溶解度参数越接近,表明分子间的相容性越好,分子在溶液体系中越不易分相。
对于由50个相同模型分子构成的无定形晶胞体系,在373.15 K下进行动力学模拟,并在体系平衡后进行内聚能密度计算,将计算结果取平方根后即得到该种物质的溶解度参数。为验证上述溶解度参数计算方法的可靠性,采用相同的模拟方法对3种已知小分子进行密度(ρ)、溶解度参数的模拟计算,结果如表1所示。
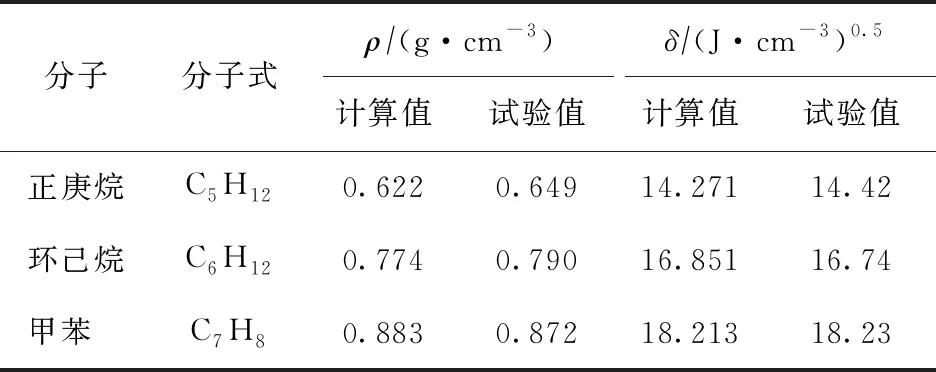
表1 3种小分子的密度、溶解度参数模拟计算结果[21]
由表1可知,由上述模拟得到的计算值与试验值[21]相近,表明该模拟计算方法具有良好的可靠性。进一步计算沥青质分子及柴油原料分子的溶解度参数,结果如表2所示。
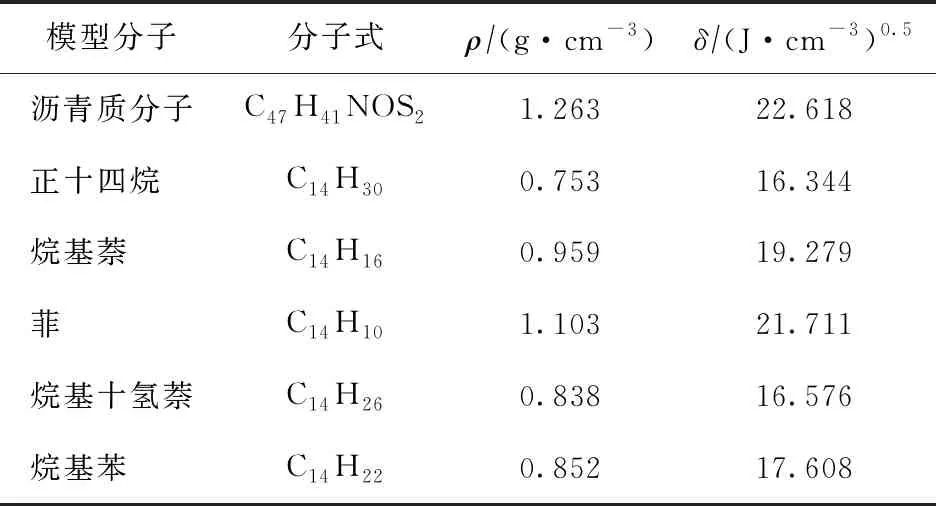
表2 模型分子密度、溶解度参数模拟计算结果
由表2可知,不同结构分子的溶解度参数各不相同,其中沥青质分子的溶解度参数最大,为22.618(J·cm-3)0.5,与之较为接近的是LCO原料中的菲和烷基萘分子,而正十四烷与加氢柴油原料中的烷基十氢萘、烷基苯分子的溶解度参数最为接近,为16~18(J·cm-3)0.5。同时,正十四烷、烷基十氢萘以及烷基苯分子的溶解度参数与沥青质分子相差较大,表明这3种分子与沥青质分子的相容性较差。结合上述分子结构中的静电势分布及单分子间的结合能分析,菲与烷基萘在结构上与沥青质分子较为相似,其分子间因π电子云重叠能形成较强的π-π相互作用,因而其在混合时更容易互溶。饱和烃分子与沥青质分子存在明显的结构差异,其分子间仅有较弱的范德华作用,二者混合时更倾向于分相;而烷基苯因其分子结构中较长烷基侧链的影响,其芳环结构不易接近沥青质分子,二者混合也存在相分离的趋势。
2.4 二元模拟溶液
通过单分子性质的模拟计算,对重质船燃分子结构特征、分子间作用强弱及互溶性有了深入的了解。然而,溶解度参数仅可用于对不同结构重质船燃模型分子间互溶性的预测,并不能真实反映分子在溶液中的相行为。同时,上述研究还发现,沥青质分子间具有较强的相互作用,可能会在体系中聚集,因此在重质船燃调合过程中沥青质组分在整个体系中的相行为更受关注。基于此,构建了以沥青质分子为溶质分子、其他某种模型分子为溶剂分子的二元模拟溶液模型,进一步在373.15 K、标准大气压下探究沥青质与不同柴油组分的互溶性,考察沥青质分子在柴油溶液中的分子分布。
2.4.1沥青质模型
在构建二元溶液模型前,为模拟计算不同大小沥青质分子模型体系的动力学特性,分别构建了分子数为10,20,30,40,50的5个沥青质分子一元体系模型,并分别对其结构进行优化并退火。经动力学模拟体系平衡后,对5个沥青质分子模型体系分别进行密度、溶解度参数、内聚能的计算,结果如表3所示。
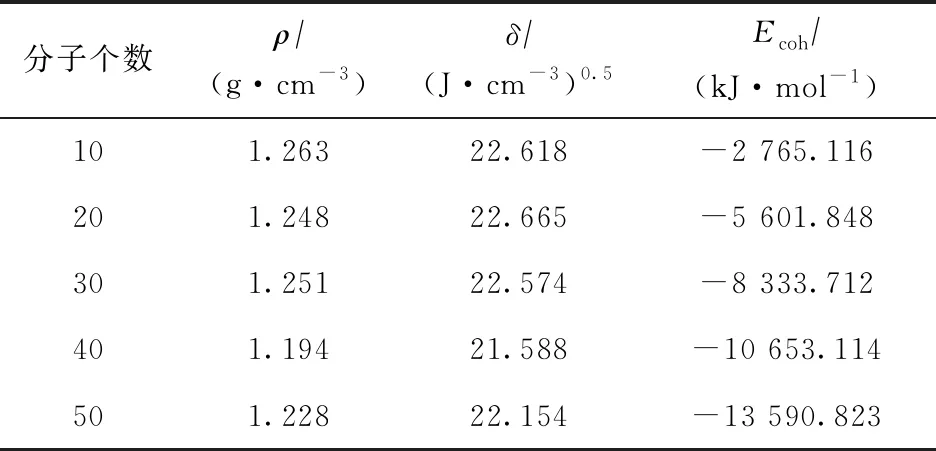
表3 不同大小沥青质分子一元模型体系性质的计算结果
各沥青质分子模型体系平衡时,沥青质分子的分子分布如图6所示(隐藏了晶格)。从截取的平衡轨迹图像可知:沥青质分子间具有较强的聚集倾向,模型中形成了尺寸较大的多聚体结构,聚集体主要以π-π堆叠形貌为主;而且,随着沥青质分子数量增多,π-π堆叠尺寸逐步增大,沥青质聚集体的内聚能也逐渐增大,这表明内聚能可以反映沥青质的团聚程度。
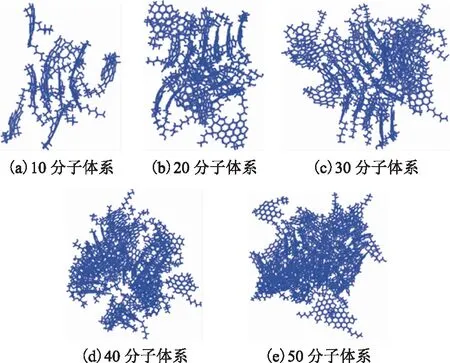
图6 沥青质分子一元模型体系的分子分布
2.4.2二元溶液模型的动力学模拟
将沥青质分子分别与正十四、烷基十氢、烷基苯、菲的分子混合,构建二元模拟体系,并调整溶剂的质量分数(下同)分别为30%,50%,70%,考察分子间作用强弱与溶剂含量对沥青质分子在溶液中相行为的影响。截取12个溶液模型动力学模拟平衡时的轨迹图像,分别如图7所示。
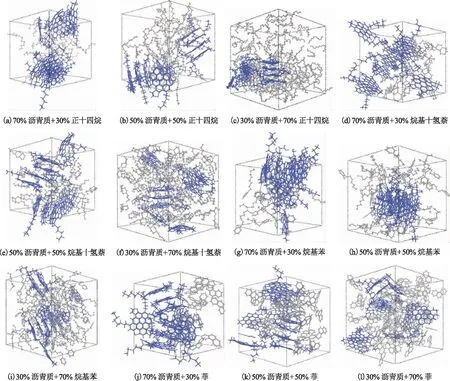
图7 含沥青质分子与柴油组分分子的二元溶液体系动力学模拟平衡时的轨迹图像
由图7可知,对于沥青质-饱和烃溶液模型,饱和烃分子正十四烷、烷基十氢萘与沥青质分子不相溶,模拟体系中沥青质分子发生聚集形成了尺寸较大的聚集体;即使增加饱和烃的量,体系中的沥青质分子依旧发生明显的聚集,并且有自成一相的趋势,表明沥青质分子在饱和烃中的分散性较差。而在沥青质-菲溶液模型中,当菲含量较低时,模型体相中存在沥青质分子的多聚体,但其聚集尺寸相较于沥青质-饱和烃体系较小;增加体系中菲的量,体相中聚集体的尺寸明显减小,仅局部存在沥青质分子的二聚体或三聚体,其他沥青质分子以单分子的形态分散于体相中。对于沥青质-烷基苯溶液模型,当长链烷基苯溶剂的质量分数升至70%时,体相中仍存在尺寸较大的沥青质聚集体,说明沥青质分子在长链烷基苯中的分散性比在沥青质-菲溶液体系差。
结合模型分子的模拟计算结果可知:饱和烃分子与沥青质分子间的相互作用弱于含芳环结构烃分子与沥青质分子间的相互作用,导致沥青质分子不能很好地被饱和烃溶剂分子胶溶分散;芳香烃分子与沥青质分子虽然具有良好的相互作用,但当芳香烃量较少时,其无法很好地胶溶分散沥青质分子,同时含有长烷基侧链的芳香烃分子对沥青质分子的胶溶分散效果也不佳。这说明,当分子间相互作用较弱时,二元溶液体系中两种组分不相溶且有明显的相分离趋势;当分子间相互作用较强时,若溶剂分子足够多,就能很好地胶溶分散极性沥青质分子。
对溶剂质量分数均为70%的沥青质-正十四烷、沥青质-菲、沥青质-烷基十氢萘、沥青质-烷基苯二元溶液模型进行RDF分析,对比沥青质分子周围出现溶剂分子概率的大小,结果如图8所示。
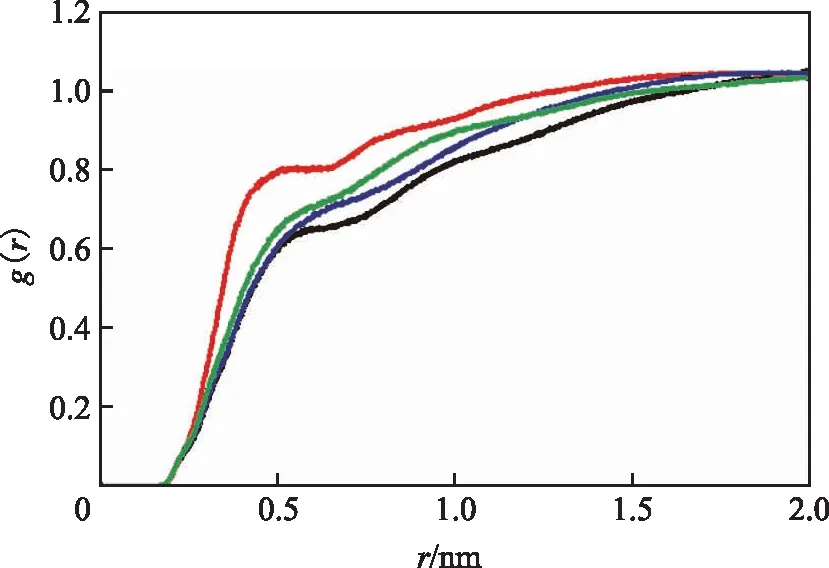
图8 4种二元溶液模型体系的RDF对比
由图8可知,在相同半径下,沥青质-菲体系的RDF最大,沥青质-烷基苯体系次之,沥青质-饱和烃体系最小,表明芳环结构分子更容易接近沥青质分子,而且芳环数越多、烷基侧链越短,则在沥青质分子周围出现的概率越大。此外,沥青质-菲曲线最大RDF峰值对应的半径为0.329 nm,依据相关研究[23-24],该半径在含芳环组分π-π相互作用范围内。图9为不同模型分子间堆叠形式和相互作用示意。如图9所示:菲分子与沥青质分子形成了面对面式的π-π堆叠相互作用;烷基苯分子也与沥青质分子形成了π-π堆叠相互作用,但其长烷基侧链不利于其与沥青质分子接近,导致其对应的RDF峰值比菲小;饱和烃分子仅能与沥青质分子的烷基侧链发生较弱的范德华作用,在模拟溶液体系中与沥青质分子距离较远。
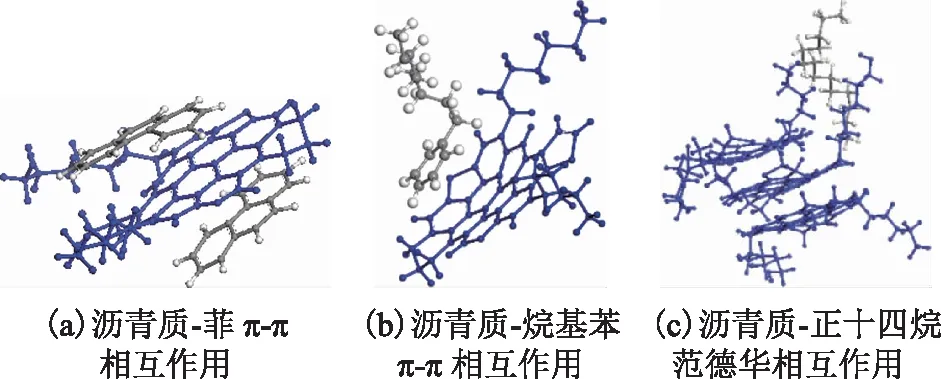
图9 模型分子间的相互作用
RDF分析可以对溶液中的分子分布进行定性分析,而对模拟溶液中分子分布的量化分析则需计算不同组分之间作用能。组分间的作用能既包含单分子间的作用能,也要结合分子数量的影响,从而反映出模拟溶液中组分间的相溶性情况。二元模拟溶液体系中沥青质与溶剂组分间的作用能计算结果如表4所示。
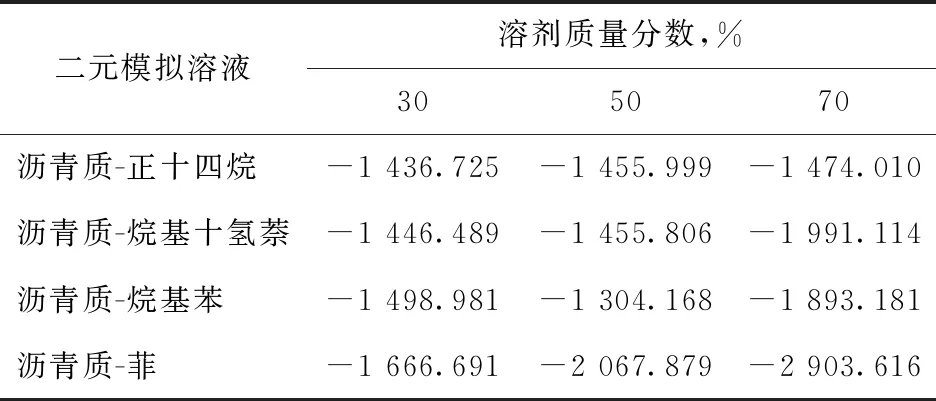
表4 不同模拟溶液中组分间的作用能模拟计算结果 kJ/mol
由表4可知:当溶剂含量较低时,组分间作用能在较低水平;随着溶剂含量升高,不同二元体系的组分间作用能均有增长。相比于沥青质-菲溶液体系,饱和烃分子溶液中的组分间作用能增幅较小,即使饱和烃质量分数达到70%,组分间作用能仍维持在较低水平;沥青质-烷基苯溶液体系中的组分间作用能也相对较小,可能是因为烷基苯中长烷基侧链不利于其与沥青质分子间发生相互作用。随着沥青质-菲溶液体系中菲含量的增加,其组分间作用能显著升高。
将以上二元溶液体系组分间作用能与沥青质一元体系内373.15 K下的内聚能(-2 765.116 kJ/mol)进行比较,可以发现:在沥青质-饱和烃、沥青质-烷基苯二元溶液体系中,即使溶剂质量分数达到70%,其组分间作用能仍比沥青质的内聚能小;而在沥青质-菲二元溶液体系中,当溶剂质量分数达到70%时,组分间作用能大于沥青质的内聚能。由于组分间作用能与沥青质内聚能的相对大小和体系中分子分布有较好的对应关系,由此可知饱和烃、烷基苯难以胶溶分散沥青质,而质量分数70%的菲作为溶剂能够很好地胶溶分散沥青质。
3 结 论
(1)不同来源的重质船燃分子,因其分子结构不同致使其电势分布各不相同,从而导致不同分子间的相互作用也各不相同。柴油调合原料中的烷基苯、烷基萘、菲等芳香烃分子结构与重质船燃中沥青质组分分子的结构较为相似,因而其与沥青质分子间能形成较强的π-π相互作用,分子间结合能较高、溶解度参数相近;饱和烃分子与沥青质分子结构差异较大,其分子间仅有较弱的范德华力作用,二者的溶解度参数相差较大。
(2)采用MD方法对沥青质-溶剂二元溶液体系模拟计算发现:菲与沥青质组分间相互作用较强,当其在二元体系中质量分数达70%时,可使沥青质胶溶分散且不易相互聚集;含长烷基侧链的烷基苯因侧链的影响,导致其与沥青质组分间作用能大幅减小,不利于对沥青质分子的胶溶分散;链烷烃或环烷烃均与沥青质分子作用较弱,即使含量很高,也无法胶溶分散沥青质分子。
(3)373.15 K下沥青质的内聚能为-2 765.116 kJ/mol。为使沥青质胶溶分散,不仅溶剂与沥青质组分间的相互作用要强,溶剂含量也必须足够高。原因在于:组分间作用能随着溶剂含量的增加而增大,当组分间作用能大于沥青质内聚能时,才能实现对沥青质的胶溶分散,且二者差值越大,沥青质-溶剂的胶溶分散体系越稳定。
猜你喜欢
化工设计通讯(2022年9期)2022-10-10
石油沥青(2021年2期)2021-07-21
绿色中国(2019年17期)2019-11-26
西南石油大学学报(自然科学版)(2018年6期)2018-12-26
天然气与石油(2015年2期)2015-02-28
食品工业科技(2014年15期)2014-03-11
纯碱工业(2014年6期)2014-03-11
华东理工大学学报(自然科学版)(2014年3期)2014-02-27
公务员文萃(2013年5期)2013-03-11
中国洗涤用品工业(2012年4期)2012-03-20
