
基于BSA-seq和RNA-seq挖掘水稻株高相关QTL
2023-09-01 10:41吴元明林佳怡柳雨汐李丹婷张宗琼郑晓明逄洪波
生物技术通报 2023年8期
吴元明 林佳怡 柳雨汐 李丹婷 张宗琼 郑晓明 逄洪波
(1. 沈阳师范大学生命科学学院,沈阳 110034;2. 广西壮族自治区农业科学院水稻研究所 广西水稻遗传育种重点实验室,南宁 530007;3. 中国农业科学院作物科学研究所,北京 100081;4. 海南三亚中国农业科学院国家南繁研究院,三亚 571700;5. 国际水稻研究所,菲律宾马尼拉 DAPO box 7777)
株高是影响水稻(Oryza sativa L.)产量的重要农艺性状之一,自“绿色革命”的兴起[1-2]以及与株高相关代表基因sd1[3]和Rht[4]被发现并阐明作用机制后,形成了对株高与产量之间关系的正确理解:适度的植株矮化增强了其抗倒伏性,使产量更加稳定。普通野生稻(Oryza rufipogon Griff.)是亚洲栽培稻的祖先,广泛分布于全球各地,遗传多样性丰富,具有较强的适应性和抗逆能力,是栽培稻品种改良和培育的重要种质资源库。染色体片段代换系(chromosome segment substitution lines, CSSLs)群体作为分子育种研究中常用的试验材料之一,其主要特点是来源于亲本的单个染色体段被替换成轮回亲本背景,即每一个CSSL除了携带1个或少数几个来自供体亲本的代换片段外,其遗传背景均和轮回亲本一致,能有效简化遗传背景,是研究QTL的重要群体之一。
随着现代生物技术的发展,越来越多与株高性状相关的基因与数量性状基因座(quantitative trait locus, QTL)被成功定位。如Liu等[5]在水稻第5染色体定位的抗倒伏基因SBI,该基因编码GA-2氧化酶,通过抑制赤霉素(gibberellins, GAs)的活性控制水稻株高,遗传分析表明,SBI是控制水稻抗倒伏能力的半显性基因,其等位基因表现出一系列不同的产物活性。Tu等[6]通过构建RILs(recombinant inbred lines)在一个主效QTL-qPND1内定位到编码细胞分裂素(cytokinin, CTK)氧化酶的候选基因OsCKX2/Gn1a,该基因第3外显子中11 bp的缺失使得带有不同亲本型等位基因个体的抗倒伏能力表现出极显著的差异,后续研究发现抗倒伏型gn1a与SCM2[7]之间的相互作用可进一步增强水稻的抗倒伏能力。最新发现的OsFBK4[8]和DHT1[9],前者编码具有F-box和kelch repeat结构的蛋白,通过抑制GAs活性进而促进矮化,后者的产物参与构成剪接体使独脚金内酯(strigolactone, SL)受体基因D14正常表达,D14促进SL信号抑制物D53的降解,令SL正常发挥作用,进而抑制分蘖形成,其突变形式dht1使D14的mRNA剪接受阻,SL无法发挥作用,进而促进分蘖形成以促进矮化。这一系列研究发现株高的本质是受一系列基因与环境因素共同制约的数量性状[10-11]。GA[12]和油菜素内酯(brassinolide,BR)[13]是已知的参与株高调控过程的关键成员,后续发现SL[14]和生长素(auxin, IAA)[15]也参与这一过程。
虽然已经在水稻中鉴定到一些与株高相关的QTL和相关基因,但相比于株高这个复杂的数量性状而言,对于这些基因的功能和调控机制的理解还不充分。需要进行更多的研究来挖掘株高基因,以充分理解它们在株高调控中的作用。本研究使用籼稻油占8号和广西普通野生稻构建的CSSL群体以及现代分子生物学技术,对广西普通野生稻进行研究,通过二代测序(next generation sequencing, NGS)和混池分析(bulk segregation analysis, BSA)方法,鉴定广西普通野生稻基因组中与株高相关的QTL,进而筛选出在株高调控中可能起到关键作用的候选基因。同时,运用RNA-seq分析进一步验证这些候选基因在株高调控过程中的表达量与水平,为进一步探究水稻株高调控机制提供了重要的理论指导和研究思路,为培育理想株型的水稻提供理论依据。
1 材料与方法
1.1 材料
以广西普通野生稻(Oryza rufipogon Griff.)(Guangxi common wild Rice, GCWR)和籼稻油占8号(Oryza sativa L.)(Youzhan8, YZ8)分别作为供体和受体,按照图1所示流程,采用单粒传法构建得到285个CSSL群体,由广西壮族自治区农业科学院水稻研究所提供。

图1 本研究所用的回交群体的构建过程Fig. 1 Construction process of the backcross populations used in this study
1.2 方法
1.2.1 株高表型鉴定与极端性状混池的构建 每个CSSL群体随机选取20粒种子种植于海南省三亚市国家南繁研究院的试验田中,亲本与子代各群体均随机选取5株用于测量抽穗期株高。分别选取最高株和最矮株的20个系构建高株池(H-pool)和矮株池(L-pool),收集其叶鞘和节间组织,液氮快速冷冻后,提取其基因组DNA和总RNA。
1.2.2 总DNA的提取及NGS测序 采用CTAB法从每个极端群体提取总量为1.5 μg的基因组DNA作为构建文库的原料,按照生产商提供的标准使用Truseq Nano DNA HT Sample preparation Kit(Illumina USA)构建测序文库。大致流程为:总DNA由超声波处理形成长度为350 bp的片段,对其进行末端修复、3'末端添加A尾、连接接头等处理用于后续的Illumina测序和PCR扩增。PCR产物在AMPure XP system中进行纯化,文库经Agilent2100 Bioanalyzer和real-time PCR完成定量分析。经上述方法构建完成的文库在Illumina HiSeq 4000 platform中完成测序,最终生成全长为350 bp的双端测序片段。
1.2.3 测序数据质控及比对 原始测序数据需要经过一系列质量控制程序的处理,去除人为误差后可用于参考基因组比对和后续分析。质量控制标准包含以下3个方面:(1)去除未知碱基含量≥10%的片段;(2)去除碱基质量值<5的片段;(3)去除与接头互补核苷酸>10 nt的片段,该过程允许的最高错误匹配几率为10%。
将上述质控处理后的样品测序数据用Burrows-Wheeler Aligner(BWA)[16]软件对比至Nipponbare(NIP)基因组中(settings: mem -t 4 -k 32 -M -R),参考基因组MSU 7.0下载自http://rice.plantbiology.msu.edu/pub/data/Eukaryotic_Projects/o_sativa/annotation_dbs/pseudomolecules/version_7.0/all.dir/。SAMtools software[17]用于将比对结果转化为bam文件(settings:-bS -t)。
1.2.4 基于NGS测序的BSA-seq分析与候选基因挖掘 GATK[18]软件中的Unified Genotyper function用于完成对各样品SNP和InDel的检测,并分别使用GATK中不同的参数对SNP(settings: --cluster-WindowSize 4, --filter Expression “QD<4.0||FS>60.0||MQ<40.0”,-G_filter“GQ<20”)和InDel(settings:--filter Expression “QD<4.0||FS>200.0||Read PosRankSum<-20.0||Inbreeding Coeff<-0.8”)进行过滤以防止低质量和无效的分子标记对后续分析造成影响。用ANNOVA[19]将过滤后的SNP和InDel在参考基因组文件中进行注释。
以日本晴基因组MSU 7.0作为参考,比对子代池中与参考基因组在特定碱基位点相同或不同的reads数量,计算不相同reads数量占总数的比例,即为SNP-index。InDel-index的计算方法与SNPindex基本相同,只是InDel涉及长度不等的DNA片段而非单个碱基。若两个子代池中出现SNP/InDelindex<0.3的位点,则将其过滤掉,以减少测序和比对误差造成的影响[20]。选用1 Mb为窗口大小、1 kb为步长作为滑动窗口方法的标准参数,并用该方法对SNP/InDel-index在染色体上的分布进行作图。计算2个子代池中的SNP/InDel-index的差值,结果即为Δ(SNP/InDel-index)。SNP/InDel-index关联分析所需的统计学置信区间按照标准方法设置为95%和99%。根据计算的SNP/InDel-index和Δ SNP/InDelindex,挑选出SNP/InDel-index在2个池中差异较大,Δ SNP/InDel-index在一定置信水平下的窗口即为候选区间。在候选区间中挑选出SNP/InDel index在一个池中为1,另一个池中为0的SNP/InDel作为原始的候选SNP,根据这些SNP的注释信息,挑选在外显子上,且是非同义突变、stop gain、stop loss等重要突变的SNP,这些SNP作为候选SNP,这些SNP所在的基因作为候选基因。
使用BLAST方法将候选区间内的编码基因在GO、KEGG、Uniprot、NOG数据库中进行详细注释,以注释结果为基础对候选基因进行筛选,初步确定参与水稻株高调控的候选基因。
1.2.5 转录组数据分析 根据水稻GH998已有的水稻抽穗期株高转录组数据,对高株和矮株的差异表达基因进行分析,所用的转录组数据库从NCBI数据库中下载(GH998 MU, SRR16686933-SRR16686935和GH998 WT, SRR16686936, SRR16686938,SRR16686939),不同版本的基因ID编号借助The Rice Annotation Project Database[21]中的ID Converter功能完成,利用TBtools[22]软件从RNA-seq原始数据中提取株高差异表达基因并绘制热图。
1.2.6 总RNA的提取和候选基因表达模式的验证 TRIzol法提取叶鞘和节间组织总RNA,按照如下步骤完成质量检测:(1)1%琼脂糖凝胶电泳检测总RNA是否存在降解和污染;(2)NanoPhotometer® spectrophotometer(IMPLEN, CA,USA)检测纯度;(3)Qubit® RNA Assay Kit in Qubit® 2.0 Flurometer(Life Technologies, CA, USA)测定RNA浓度;(4)RNA Nano 6000 Assay Kit of the Agilent Bioanalyzer 2100 system(Agilent Technologies,CA, USA)检测RNA样品的完整性。
随机六碱基引物和M-MuLV反转录酶(New England Biolabs, Ipswich, MA)用于合成cDNA的第一链,DNA聚合酶I和RNase H合成cDNA的第二链。Invitrogen试剂盒完成双链cDNA的末端修复、加尾和接头添加。选用肌动蛋白作为内参基因。所有用于RT-qPCR的引物均采用Primer 3(https://primer3.ut.ee)软件设计(表1)。各基因设置3次生物学重复以减少误差。相对表达模式经由2-ΔΔCt方法[23]计算获得。

表1 用于RT-qPCR分析的引物列表Table 1 List of primers for RT-qPCR analysis
1.2.7 数据分析 将测量得到的CSSL群体株高求平均值分析分布频率,用Student's t-test检验RTqPCR结果的差异显著性,用GraphPad Prism v8.4.2软件绘制图形。
2 结果
2.1 株高性状评估
通过对285个CSSL群体平均株高表型进行鉴定(图2)。所有CSSL群体的平均抽穗期株高分布范围为101.9-128.2 cm,GCWR抽穗期株高为127.4 cm,YZ8抽穗期株高为103 cm。频率分布表明,株高在CSSL群体中的分布基本符合正态分布的特征,即满足数量性状的定义,因此,这些CSSL群体可用于BSA-seq分析。

图2 CSSL群体的株高表型分析Fig. 2 Plant height phenotype analysis of CSSL populations
2.2 NGS数据质控与比对
亲本与极端性状子代池的基因组DNA经Illumina平台测序后(表2),供体亲本GCWR和受体亲本YZ8分别产生11.92 Gb(Q20≥97.75%,Q30≥93.72%,GC含量为44.23%)和11.13 Gb(Q20≥97.57%,Q30≥93.31%,GC含量为44.6%)的原始数据。2个子代池H-pool和L-pool分别产生14.38 Gb(Q20≥97.34%,Q30≥92.92%,GC含量为46.54%)和14.27 Gb(Q20≥97.1%,Q30≥92.45%,GC含量为44.38%)的原始数据。每组样品的测序数据经整理和过滤后至少有97.89%成功比对至日本晴(Oryza sativa var Nipponbare)基因组MSU 7.0中。各样本的测序数据量足够,质量合格,所有样本的比对结果正常,可用于后续分析。

表2 用于BSA-seq分析的NGS测序数据统计概况Table 2 Summary of NGS data for BSA-seq analysis
2.3 基于NGS的BSA-seq数据分析
SNP-index和InDel-index两种算法被用于鉴定与株高相关的基因组区域。来自H-pool与L-pool的测序数据分别经BWA、GATK和ANNOVAR比对、分子标记检测和注释后,共得到3955 417个SNP和571046个InDels,过滤后得到13362个SNP(其中包含1088个非同义SNP)和1746个InDels。
利用Δ(SNP-index)算法在Chr.7和Chr.10上分别鉴定到3.205(1-3205 000 bp)和1.311 Mb(3782 001-5093 000 bp)的候选基因组区域(图3)。根据Δ(InDel-index)算法,在Chr.7和Chr.10上分别鉴定到大小为2.848(1-2848 000 bp)和1.292 Mb(3796 001-5088 000 bp)的候选基因组区域(图4)。值得注意的是,利用Δ(InDel-index)算法得到的候选区间完全包含在Δ(SNP-index)算法鉴定到的候选区间内。上述所有候选基因组区域的置信阈值均大于99%(P<0.01)。为了缩小定位株高相关的基因组区域,将这两种算法所得到的候选区间的共有部分作为最终的关联区段,相关信息如表3所示,分布于第7和第10染色体上的最终区段长度分别为2.848和1.292 Mb,共包含276个已注释的基因。值得注意的是,在Chr.7的候选区段中包含有已报道的与水稻株高调控相关的基因LOC_Os07g05900(PROG1)和LOC_Os07g05720(OsTCP21)。
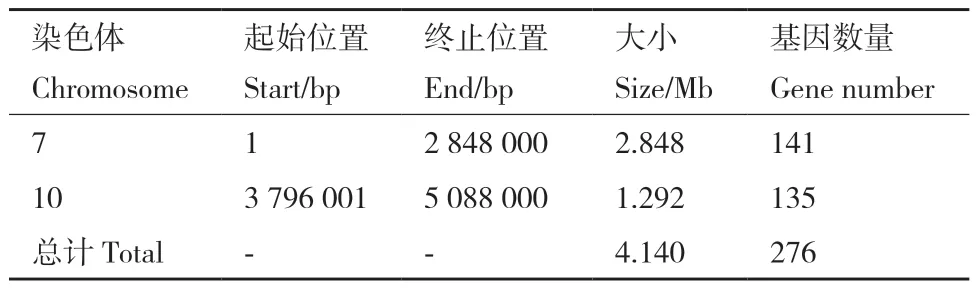
表3 Δ(SNP-index)和Δ(InDel-index)定位与株高相关的候选基因组区段Table 3 Candidate genome regions related to plant height mapping by Δ(SNP-index)and Δ(InDel-index)
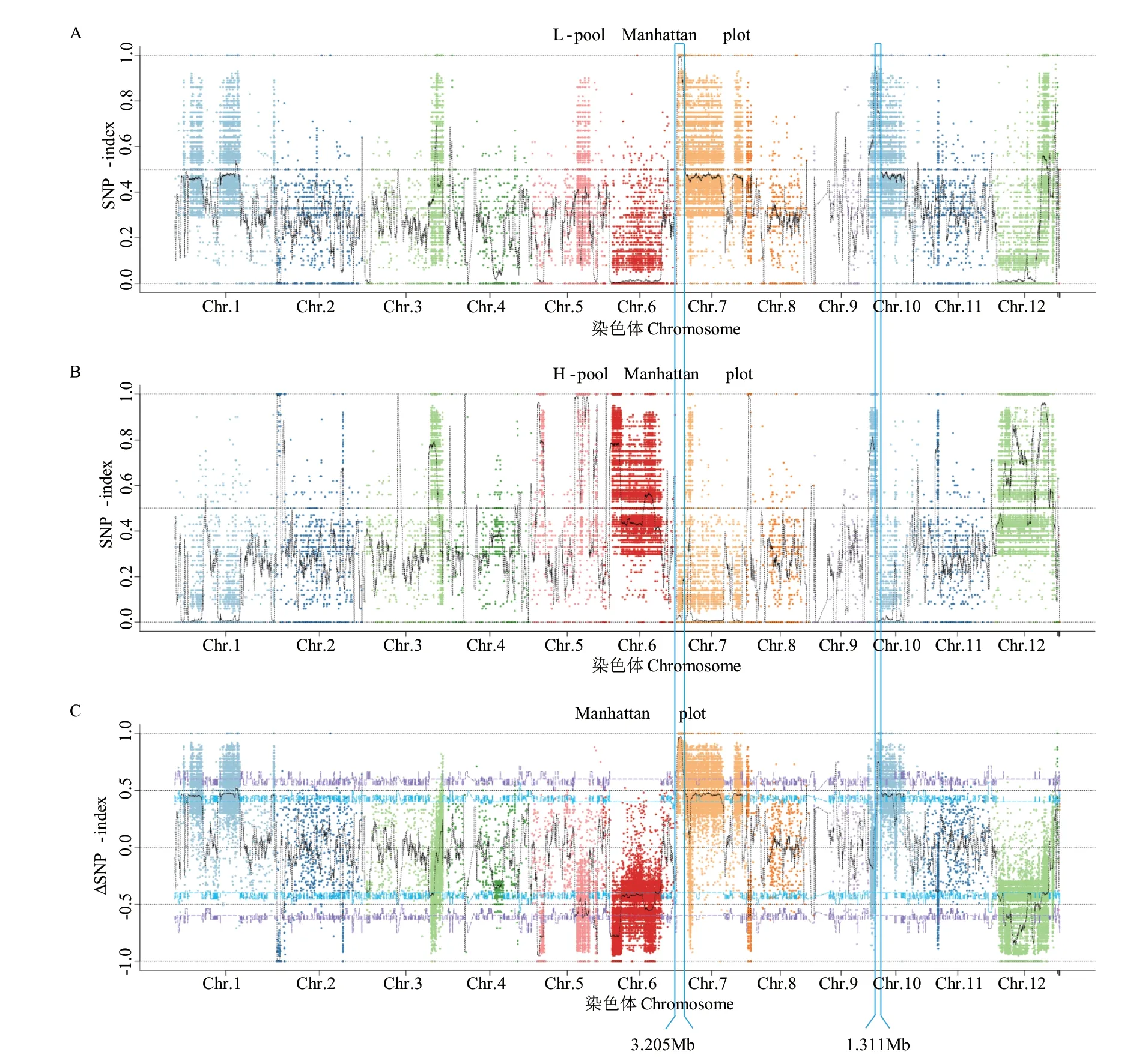
图3 基于BSA-seq分析的矮株(A)和高株(B)的SNP-index图以及Δ(SNP-index)图(C)Fig. 3 SNP-index graphs of bulk-L(A)and bulk-H(B), as well as Δ(SNP-index)graph(C)based on BSA-seq analysis

图4 基于BSA-seq分析的矮株(A)和高株(B)的InDel-index图以及Δ(InDel-index)图(C)Fig. 4 InDel-index graphs of bulk-L(A)and bulk-H(B), as well as Δ(InDel -index)graph(C)based on BSA-seq analysis
2.4 候选区间内基因的功能注释与富集分析
根据候选基因组区间内SNP、InDel调用和注释的结果,共有413个高质量的非同义突变SNP(4个nsSNP被定位在日本晴参考基因组的Chr.10内,其余nsSNP位于Chr.7内)包含在92个基因内(3个基因位于参考基因组的Chr.10内,其余基因位于Chr.7内),139个高质量InDels(125个属于上游类别,其余被归类于移码突变)被定位到85个基因内(除了位于Chr.10内的1个基因外,剩余基因全部位于Chr.7内)。将利用两种分子标记所关联到的可能的候选基因列表取交集,得到78个共有基因,这些基因即为与株高潜在相关的候选基因。
将78个基因在GO、KEGG、Uniprot、eggNOG数据库中进行功能注释以缩小候选基因的选区范围(表4)。在所有注释到的高质量nsSNPs和upstream类SNP中分别有15个nsSNP和20个upstream类SNP都位于相同的5个基因内,这些位点的Δ(SNPindex)均等于1。此外,这5个基因还包含有6个Δ(InDel-index)=1的upstream类InDels,其具体的功能注释信息如表5所示。LOC_Os07g02770在参考基因组中被注释为含有F-box结构域的蛋白,这是一类广泛分布于真核生物中的蛋白,具有丰富的多样性,以参与泛素化蛋白酶体途径为主要功能。LOC_Os07g02850被注释为含DUF538结构域的蛋白,该类蛋白为植物所独有,在双子叶植物与单子叶植物中均有分布。目前,已鉴定到相当数量的DUF538类蛋白,但其具体的生物学功能仍然未知。LOC_Os07g04220被注释为与水稻中唯一的红光和远红光感受器——光敏色素信号通路相关的受体类激酶。LOC_Os07g05050被注释为含有DEAD-box结构域的蛋白,其广泛分布于动物与植物中,在植物中的已知主要功能是参与对多种胁迫因素的响应过程。LOC_Os07g05720的产物是功能和作用机制已知的OsTCP21转录因子蛋白。

表4 候选区间内基因的功能注释结果统计Table 4 Statistics of functional annotation results for genes in candidate intervals
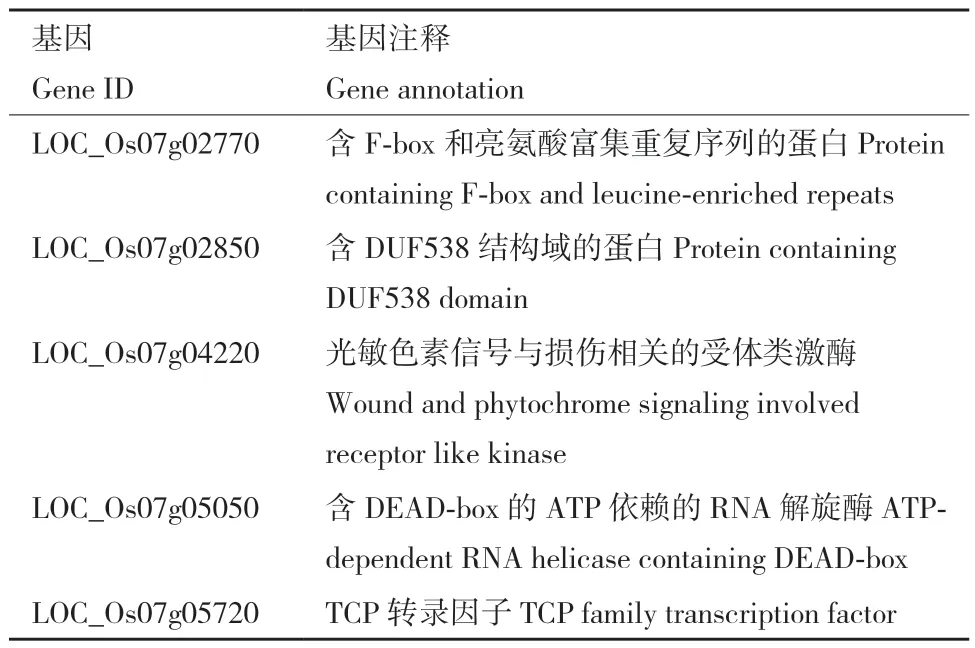
表5 候选基因功能注释结果Table 5 Results of functional annotation of candidate genes
2.5 关联区间内候选基因表达模式分析与验证
对已有的株高转录组数据(GH998MU_S和GH998WT_S)进行分析,结果(图5)表明,两种分子标记筛选到的78个共有基因中,有54个在转录组数据中可以找到。其中,编码F-box蛋白的LOC_Os07g02770(Os07g0118900)和编码OsTCP21转录因子的LOC_Os07g05720(Os07g0152000)在转录组数据中呈现差异表达,而剩余3个基因在转录组数据中表达量较低且无明显差别。
为了检验候选基因的表达模式,利用两种极端性状个体的总RNA进行RT-qPCR试验。结果(图6)显示,5个基因的表达模式在两种个体中均呈差异性表达,表明它们在株高调控过程中均具有一定的功能。3个基因(LOC_Os07g02770、LOC_Os07g04220和LOC_Os07g05720)在矮株中的表达量上调,说明这些基因促进了矮株的形成。2个基因(LOC_Os07g02850和LOC_Os07g05050)则在高株个体中表达量更高,表明其参与株高的正向调控。此外,在5个用于RT-qPCR验证试验的基因中,LOC_Os07g05720的产物为功能已知的TCP家族的转录因子,与miRNA互作以负向调控株高。该基因的RT-qPCR验证结果与已有研究相一致,证明本研究中BSA-seq结果的可靠性。

图6 RT-qPCR验证候选基因Fig. 6 RT-qPCR validation of selected candidate genes
3 讨论
3.1 InDels与SNPs在BSA分析方法中的应用
与质量性状相比,数量性状受到更多基因的共同制约,各基因之间相互作用的复杂程度也远大于前者。随着测序通量的不断提升,下一代测序技术的关注点由目标物种的全基因组序列转变为与数量性状潜在关联的多种分子标记。在过去的研究中,SNP标记已经成功地应用于许多研究中[24-30],因为它们易于获取和分离,并且通常具有较高的多态性。然而,随着研究深入,InDels标记作为另一种遗传变异类型,也逐渐引起了人们的关注和重视,其在BSA-seq中的应用已经逐步得到了证明,如番茄(Solanum lycopersicum L.)[31]和木豆(Cajanus cajan L.)[32]中通过关联InDels初步定位到与疾病抗性相关的基因组区域。与SNP标记相比,InDels标记可以提供更多的遗传信息,因为它们不仅包括碱基的改变,还包括DNA序列的插入和缺失。因此,InDels标记可以扩大连锁不平衡区域的范围,增加标记密度,并提高遗传位点的鉴定准确性。另外,由于InDels位于遗传连锁的边界区域,其类型和数量的变化也较为明显,有助于精细地调控目标基因,并为研究数量性状的遗传基础提供更全面、详尽的信息[33]。与此同时,InDels标记也存在一些局限性,比如其检测和分离的难度较大,特别是对于长度不同的InDels,并且对复杂和大基因组物种的数据集的分析需要对用于标记性状关联分析的InDels选择标准进行一些额外的修改等。
本研究使用了SNP和InDels两种分子标记共同进行BSA-seq分析。这两种分子标记分别涉及基因编码区碱基类型和数量的变化,由于密码子的简并性,SNP造成的影响可能会被抵消,而InDels所在DNA区域内碱基数量发生的变化,极易使基因功能发生改变。其次,在已有的SNP标记基础上进行InDels标记,有利于增加标记密度、合并候选区间并为开发新的InDels标记提供理论基础,而本研究结果也表明了InDels对于性状定位和功能标记开发的重要性。
3.2 候选区域中与株高相关的基因
本研究利用SNP和InDel共同关联到5个与水稻株高相关的候选基因,且全部位于Chr.7内。其中OsTCP21、LOC_Os07g05050和LOC_Os07g02850在矮株中呈下调表达,LOC_Os07g04220和LOC_Os07g02770在高株中呈下调表达。其中OsTCP21隶属于TCP转录因子,该基因已被证实与促进水稻矮化相关[34],其转录产物可被miR319靶标进而导致功能受到抑制。此外,通过深入分析发现,Chr.7的候选基因组区域中含有已报道的基因——LOC_Os07g05900(PROG1),其产物为C2H2型锌指蛋白转录因子,PROG1功能的丧失是栽培稻株型改良、得以直立生长的关键[35-37]。
株高作为决定水稻产量的重要因素,一直是遗传学和育种学研究的焦点。目前,对水稻株高的遗传调控机制的研究主要集中在几个方向。首先是关于植物激素生长素调控水稻株高的研究。近年来,越来越多的研究表明生长素识别和转导信号通路对水稻株高起着重要作用[38-39],生长素水平的调节对植物细胞伸长和分化等过程有很大影响。其次,还有一些研究致力于探究一些转录因子调控水稻株高的机制,比如TCP类[40]和bZIP类[41]转录因子等。这类转录因子可以与生长素信号通路进行交叉调控,并对细胞伸长、分化等发育方式进行调节。另外,一些关于miRNA在水稻株高调节中的作用研究表明[42],miRNA以通过调节特定基因表达活性的方式对水稻的株高进行调控。但是,以上研究大多只停留在基因表达水平的探究,对于这些基因如何影响水稻株高的机制还有待进一步探索。
本研究利用高通量测序及生物信息分析方法,筛选出与水稻株高关联的5个候选基因,这些基因均位于Chr.7内。LOC_Os07g05050编码隶属于RNA解旋酶超家族II[43]的DEAD-box蛋白,这类蛋白与真核生物的生长发育密切相关,如拟南芥(Arabidopsis thaliana)中与低温相关的LOS4[44],水稻中提升高温耐性的TOGR1[45]以及葡萄(Vitis vinifera L.)中与干旱耐性相关的VviDEADRH25a[46]。LOC_Os07g02850产物具有植物特有的DUF538结构域,已报道的参与光合作用的水溶性叶绿素结合蛋白CaWSCP[47]和水解叶绿素的羧酸酯酶[48]均具有该结构域。LOC_Os07g04220编码与光敏色素信号相关的受体类激酶,光敏色素C已被研究与玉米(Zea mays L.)株高调控相关[49],近期的一项研究中发现,光敏色素C编码基因PHYC的功能缺失突变体参与水稻株高的调节[50]。LOC_Os07g02770的产物是具有F-box结构域的蛋白,该类蛋白通过介导泛素化蛋白酶体途径来调节植物的多种生命活动[51],如拟南芥中的F-box蛋白——SAGL1突变体表现出明显的株型矮化[52]。本研究结果为进一步探究水稻株高的遗传调控机制提供了新的思路。本研究发现这些基因都位于Chr.7上,这是一个重要的发现。因为Chr.7上有很多其他具有基因功能的序列,这些序列可能也与水稻株高相关。在未来的研究中,可以将这些序列与发现的基因进行比较,以确定它们之间的关系。此外,还需进一步研究这些基因与生长素、RNA等方面的交叉互作关系,揭示更深层次的水稻株高遗传调控机制。
4 结论
位于Chr.7中的5个候选基因的差异表达可能与水稻株高性状密切相关,其中,OsTCP21可能是株高的重要调节基因,其表达受miR319靶向和下调的调控。
猜你喜欢
今日农业(2021年11期)2021-08-13
世界科学技术-中医药现代化(2020年2期)2020-07-25
农村百事通(2019年17期)2019-10-08
临床医药文献杂志(电子版)(2017年11期)2017-05-17
现代农业科技(2017年1期)2017-03-06
江苏农业科学(2016年8期)2017-02-15
江苏农业科学(2016年8期)2017-02-15
中学生物学(2016年8期)2016-01-18
遗传(2014年3期)2014-02-28
新疆农垦科技(2014年6期)2014-02-28
