
YFV17D非感染性报告复制子及假病毒包装系统的建立
2023-09-01 10:41何宇航胡涛吴震贺煜程安春陈舜
生物技术通报 2023年8期
何宇航 胡涛 吴震,3 贺煜,3 程安春,3 陈舜,3
(1. 四川农业大学动物医学院禽病防治研究中心,成都 611130 ;2. 四川农业大学动物医学院预防兽医研究所,成都 611130;3. 动物疫病与人类健康四川省重点实验室,成都 611130)
黄热病毒(yellow fever virus, YFV)属于黄病毒科(Flavivirdae)黄病毒属(Flavivirus),该类属成员还包括昆津病毒(Kunjin virus, KUNV)、坦布苏病毒(Tembusu virus, TMUV)、登革热病毒(Dengue virus, DENV)和日本脑炎病毒等。与其他黄病毒属成员一样,YFV为单股正链RNA病毒,基因组全长11 kb左右,拥有一个开放阅读框(open reading frame, ORF)。ORF编码一个多聚蛋白,包括3种结构蛋白:衣壳蛋白(capsid protein, C protein)、前膜蛋白(premembrane, prM protein)和囊膜蛋白(envelope protein, E protein),以及7个非结构蛋白(non-structural protein): NS1、NS2A、NS2B、NS3、NS4A、NS4B和NS5。在ORF两侧为两个高度结构化的非编码区(untranslated region, UTR)5' UTR 和3' UTR,包含病毒RNA合成以及影响病毒翻译和复制所必要的顺式作用元件[1]。YFV有两种野生毒株,包括Asibi毒株和法国噬内脏病毒,其中YFV17D是由 Asibi毒株在猴子、小鼠和鸡细胞培养物中进行了 176 次传代而来,其保留了病毒的免疫原性,失去了噬内脏性、噬神经性毒性和在蚊子体内传播能力[2-4]。
亚基因复制子系统作为反向遗传学的重要组成部分,与感染性克隆一样能作为研究病毒强有力的工具。黄病毒亚基因复制子是保留了全部的非结构蛋白和非编码区的顺式作用元件,删除了部分结构蛋白基因的一种缺陷型基因组,它能够在宿主细胞内自行完成翻译和复制[5]。将报告基因引入以代替缺失的结构蛋白基因区域,并通过对报告基因表达的蛋白进行检测,可以更加容易监测亚基因复制子在细胞中的复制情况[6-8]。在复制子基础上,通过携带编码病毒结构蛋白(C、prM、E)的重组质粒来提供缺失的结构蛋白,与可以自我复制的病毒亚基因组产生打包复制子基因组的假病毒颗粒,其在结构上近似于野生型病毒,具有与野生病毒相似的进入、翻译和复制的过程[9-11]。但由于这种病毒样颗粒所包含的基因仍然缺乏必需的结构蛋白基因,无法再次包装成成熟的病毒颗粒,故而这种病毒样颗粒无法从宿主细胞中释放启动新一轮的感染,因此称之为单轮感染病毒颗粒(single round infectious virus particles, SRIPs)[12]。这种通过反式包装出的SRIPs已经在TMUV、DENV、KUNV和寨卡病毒上实现应用[13-16]。
本研究利用反向遗传学技术成功分别构建携带3种不同报告基因(Nluc、oxGFP和mCherry)的YFV17D复制子。通过检测Nluc萤光素酶蛋白、oxGFP和mCherry荧光蛋白在细胞中的表达和对病毒双链RNA(double strand RNA,dsRNA)染色观察,监控YFV17D复制子在BHK-21细胞中翻译和复制。将表达结构蛋白的包装质粒(pcDNA3.1-C28prME和pcDNA3.1-CprME)与复制子共同转染至BHK-21细胞中,收集上清再次感染BHK-21细胞,通过检测细胞内的Nluc活性和oxGFP、mCherry蛋白表达来监控SRIPs 的产生而成功建立YFV17D的假病毒包装系统。构建携带报告基因的YFV17D复制子,可以帮助深入了解YFV基因组翻译和复制的调控机制。同时,基于复制子的 SRIPs系统为研究病毒吸附、进入和组装,以及病毒与宿主相互作用提供了有力的工具。
1 材料与方法
1.1 材料
乳仓鼠肾细胞BHK-21由本实验室保存、携带报告基因的单拷贝pCC1-YFV17D-rep复制子质粒(爱沙尼亚塔尔图大学Andrews Merits教授惠赠)、大肠杆菌感受态细胞DH5α、Sure菌株、低拷贝质粒pACYC177和真核表达质粒pcDNA3.1均由本实验室保存;高保真 KOD OneTMPCR Master Mix购于东洋纺生物科技有限公司;限制性内切酶购于NEB公司;同源重组酶购于莫纳生物科技有限公司;核酸脂质体转染试剂购于翌圣生物科技股份有限公司;NanoLuc萤光素酶检测试剂盒购于Promega公司;鼠抗dsRNA J2 抗体购于SCICONS公司;FITC荧光和TRITC荧光标记羊抗鼠lgG购于Abcam公司;DAPI购于北京酷来搏科技有限公司。
1.2 方法
1.2.1 报告复制子和包装质粒构建 使用重叠延伸PCR技术和同源重组策略来构建YFV17D报告复制子。在pACYC177载体中选择2个限制性内切酶位点SgrA I和BstE II,在YFV17D复制子基因组中选择Kpn I、Nhe I和Xho I 3个限制性内切酶位点,用于之后的酶切连接。使用TMUV感染性克隆质粒pACNR CQW1-intron扩增巨细胞病毒(Cytomegalovirus, CMV)启动子、丁型肝炎核酶(Hepatitis D ribozyme, HDVr)和猴空泡病毒40(Simian virus 40, SV40)多聚腺苷酸(polyadenylation, poly)尾信号序列。将CMV与“5' UTR-Kpn I”片段融合成P1片段,其中P1片段覆盖“SgrAI-CMV-5' UTR -Kpn I”之间的基因序列;片段P2覆盖“Kpn I-Nhe I”之间的基因序列;片段P3覆盖“Nhe I-Xho I”之间的基因序列;将HDVr和SV40 poly(A)与“Xho I-3' UTR”融合成完整P4片段,覆盖“Xho I-BstE II”之间的基因序列。接着,将P1片段与线性化的pACYC177载体同源重组,形成P1片段的亚克隆质粒pACYC-P1,随后利用Kpn I、Nhe I和Xho I限制性内切酶将P1-P4全部片段按顺序连接在pACYC177载体上,最终完成带有3种不同报告基因的YFV17D复制子质粒的构建,分别命名为:pACYC-YFV17D-Nluc-rep、pACYC-YFV17D-mCherry-rep和 pACYC-YFV17D-oxGFP-rep以及pACYC-YFV17D-Nluc-rep-ΔGDD、pACYC-YFV17D-mCherry-rep-GDD-AAA和 pACYCYFV17D-oxGFP-rep-GDD-AAA。
包装质粒 pcDNA3.1-CprME质粒由上海生工生物工程有限公司合成。pcDNA3.1-C28prME以pcDNA3.1-CprME质粒为模板,保留 C蛋白末端28个氨基酸,扩增C28prME基因,与经过Hind III和ApI I限制性内切酶酶切后的pCDNA3.1(+)线性载体连接。
1.2.2 报告复制子和包装系统的工作验证 将BHK-21细胞接种至24孔板,待细胞达到70%-90%汇合度时,按照翌圣核酸脂质体转染试剂说明书转染YFV17D报告复制子,转染6 h后更换含1%胎牛血清的培养基,然后在37℃,5% CO2的培养箱中继续培养。
YFV17D报告复制子和包装质粒共同转染至接种BHK-21细胞的6孔板中,转染后6 h更换培养基继续培养48 h,收集上清。将BHK-21细胞接种至24孔板,待细胞长到70%左右密度,弃去培养基,将收集的上清滴加至每孔中,孵育48 h后检测。
1.2.3 NanoLuc萤光素酶活性的检测 使用NanoLuc萤光素酶检测试剂盒,收集转染YFV17D-Nanolucrep后不同时间点的BHK-21细胞和孵育含SRIPs上清的BHK-21细胞。使用Glo Lysis Buffer裂解BHK-21细胞,添加至96孔板中,Nano-Glo底物和Nano-Glo assay buffer按1∶49比例混合获得Nano Glo反应物,与96孔板中细胞裂解样品混合,在微孔板发光检测仪中检测样品。
1.2.4 荧光蛋白的检测 从24孔板中收集转染YFV17D-oxGFP-rep、YFV17D-mCherry-rep后不同时间点的细胞爬片和孵育含SRIPs上清的BHK-21细胞爬片。收集的爬片使用4%多聚甲醛固定,经PBST洗涤后,使用0.25%Triton-PBS 在4℃透化1 h,PBST洗涤;细胞核经DAPI复染;最后滴加甘油封片,置于荧光显微镜下观察。
1.2.5 间接免疫荧光试验(indirect immunofluorescence assay, IFA) 从24孔板中收集转染3种报告复制子质粒的不同时间点的细胞爬片。爬片使用4%多聚甲醛固定,经PBST洗涤后,使用 0.25% Triton-PBS 在4℃透化1 h,PBST洗涤;加入5% BSA在37℃封闭1 h,使用PBST洗涤;用鼠抗dsRNA J2 抗体作为一抗,分别以FITC荧光标记羊抗鼠lgG和TRITC荧光标记羊抗鼠lgG作为二抗孵育,细胞核使用DAPI复染;最后滴加甘油封片,置于荧光显微镜下观察。
2 结果
2.1 YFV17D报告复制子的构建
以分别携带NanoLuc、mCherry、oxGFP报告基因的YFV17D复制子pCC1-YFV17D-rep为模板。在实验室现有质粒pACYC177的基础上,通过SgrA II和BstE II限制性内切酶酶切,获得pACYC177线性载体。CMV启动子、HDVr和SV40 poly(A)序列由本实验室现有的TMUV感染性克隆质粒pACNR CQW1-intron上扩增。以pCC1-YFV17D-rep为模板,扩增覆盖YFV17D复制子全部亚基因组的4个片段,分别命名为P1、P2、P3 和P4。利用同源重组酶将4个亚基因片段与pACYC177线性载体连接,获得完整的YFV17D复制子质粒。由CMV驱动的基因组可以在细胞内直接起始转录,基因组下游添加的HDVr切割转录本,产生真实的3' 末端[17],同时,SV40 poly(A)的添加提供了转录终止信号[18]。为了证明报告基因的表达是由于亚基因复制子复制和翻译的结果,将YFV17D NS5基因进行突变,将RNA 依赖的 RNA 聚合酶结构域的 GDD 基序缺失(ΔGDD)或突变成丙氨酸(GDD-AAA)(图1),构建具有复制缺陷的YFV17D复制子,作为阴性对照。
2.2 YFV17D报告复制子特性的验证
将复制子质粒YFV17D-Nluc-rep和YF17D-Nlucrep-ΔGDD转染至BHK-21细胞中,分别在转染后的4 h、8 h、12 h、24 h、48 h、72 h收集细胞样,并检测Nluc萤光素酶活性。从转染后的4-48 h,野生型复制子的Nluc萤光素酶活性持续增加,并且在48 h到达最大值;而复制缺陷型复制子在转染细胞后,从24-72 h萤光素酶活性逐渐下降。比较野生型复制子和复制缺陷型复制子Nluc表达水平发现,在转染后的48-72 h,两者Nluc水平表现出统计学显著性差异(图2-A)。以上表明,完成首轮翻译后,缺陷型复制子不能再持续表达Nluc;而野生型复制子在转染至细胞后萤光素酶活性在持续增加,说明野生型复制子能够正常复制,Nluc的表达增加是依赖复制子在细胞内持续复制而增加的。
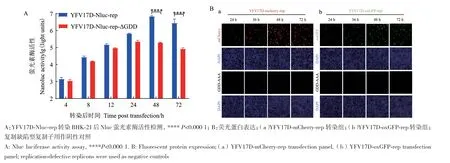
图2 YFV17D报告复制子在BHK-21细胞中的荧光蛋白表达Fig. 2 Fluorescent protein expression of the YFV17D reporter replicon in BHK-21 cells
转染携带mCherry和oxGFP荧光蛋白的YFV17D复制子至BHK-21细胞后,从转染后36-72 h,野生型复制子中的红色和绿色荧光持续增加,而复制缺陷型复制子没有可见荧光出现,两者之间呈现明显的差异(图2-B),这同样说明,荧光蛋白的持续表达需要复制子在细胞内持续复制。以上通过对不同的荧光蛋白表达的检测结果说明,我们所构建的YFV17D报告复制子能够在BHK-21细胞内正常地完成翻译和复制。
2.3 YFV17D复制子dsRNA的检测
被黄病毒感染的细胞中能检测到3种重要的RNA,包括病毒粒子RNA、双链RNA和复制中间体RNA[19]。在黄病毒基因组复制过程中,是以病毒正链RNA基因组为模板复制产生互补的负链RNA,再以合成的负链RNA为模板合成大量正链RNA[20]。由于负链RNA在整个病毒基因组复制过程中仅作为复制正链基因组的模板,且多存在于双链RNA的复制型和部分双链的复制中间体中,所以通过对dsRNA染色,可以进一步验证YFV17D复制子在细胞内的复制。将3种YFV17D报告复制子分别转染BHK-21细胞,随着转染时间的增加,3种野生型报告复制子产生的dsRNA也逐渐增加,这说明复制子在细胞内发生复制。相反,NS5突变的复制缺陷型复制子由于RNA依赖的RNA聚合酶结构的缺陷,使RNA的复制受损,无法观察到 dsRNA的产生(图3-A-C)。以上结果说明,本研究构建的YFV17D报告复制子在BHK-21细胞中具有完成自身复制的能力。
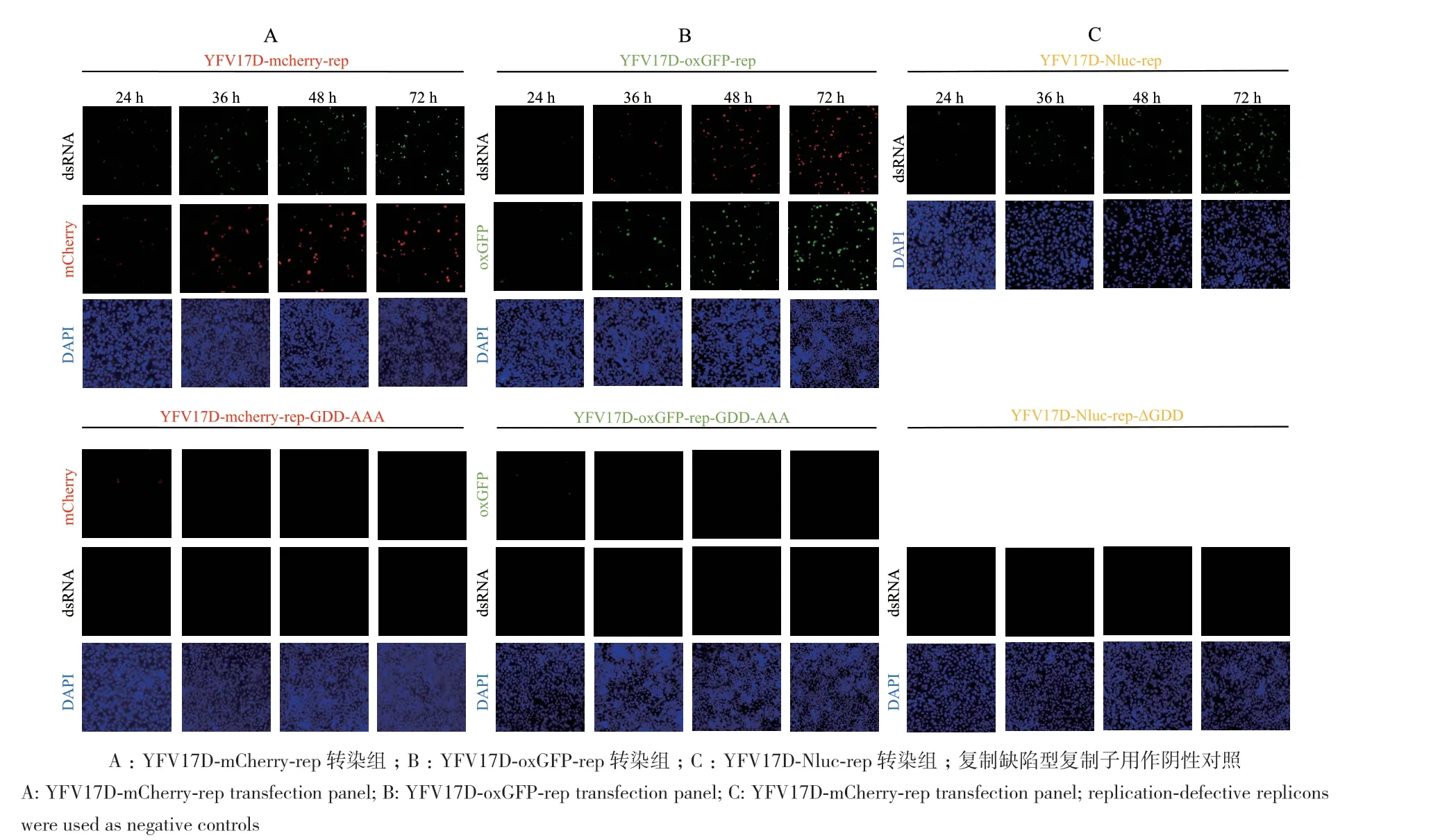
图3 YFV17D复制子在BHK-21细胞中的RNA复制情况Fig. 3 RNA replication of YFV17D replicon in BHK-21 cells
2.4 YFV17D SRIPs的产生和验证
由于复制子基因组中除了包含能够实现自身翻译和复制所需的基因元件外,缺失了大部分结构蛋白基因,而这些基因的缺失使复制子不能自主包装成具有感染能力的病毒颗粒。我们构建了表达缺失YFV17D结构蛋白的两种包装质粒pcDNA3.1-C28prME和pcDNA3.1-CprME(图4-A),并和YFV17D报告复制子质粒共同转染至BHK-21细胞中,其中将只转染报告复制子而不转染包装质粒设置为对照组。在转染后48 h,收集细胞上清,并再次感染BHK-21细胞,在上清孵育48 h以后检测相应荧光蛋白的表达水平(图4-B)。结果显示,两种包装质粒pcDNA3.1-C28prME和pcDNA3.1-CprME均可有效打包YFV17D-Nluc-rep复制子RNA,表现为荧光素酶活性显著高于对照组(图4-C);同时,两种包装质粒pcDNA3.1-C28prME和pcDNA3.1-CprME均可有效打包YFV17D-oxGFP-rep和YFV17D-mCherry-rep复制子RNA,表现为对以IFA检测到第二轮感染细胞中的绿色和红色荧光蛋白表达(图4-D)。以上结果表明,在共转染体系中,由包装质粒提供的prM和E两种结构蛋白可打包亚基因组YFV17D复制子RNA,形成具有单轮感染能力的病毒样颗粒释放到上清。
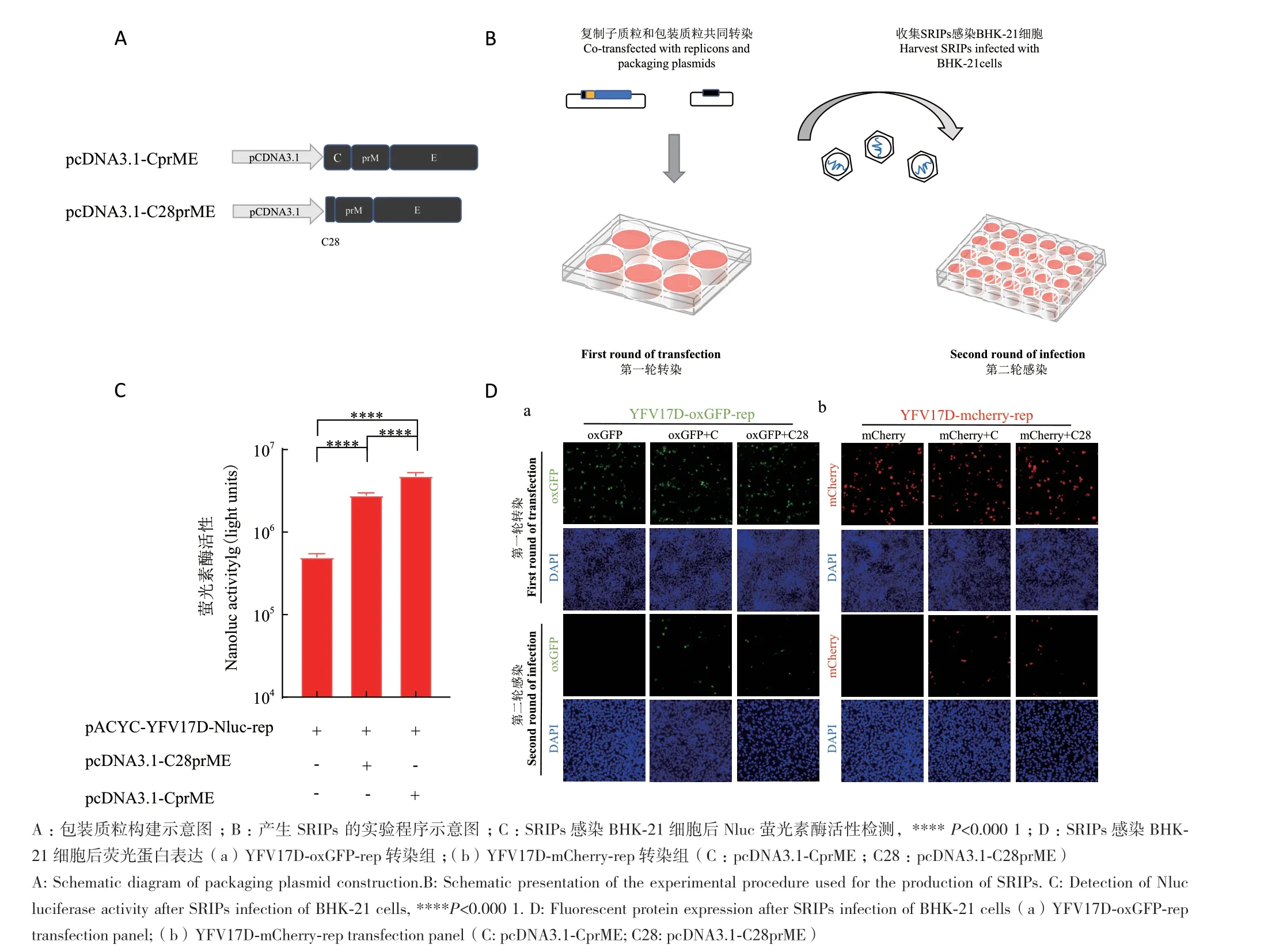
图4 YFV17D包装系统的建立Fig. 4 Establishment of the YFV17D packaging system
3 讨论
本研究以改造单拷贝质粒pCC1-YFV17D-rep为起点,旨在避免该复制子质粒以原核启动子SP6驱动起始转录的RNA转染繁琐的操作。在设计上,我们使用CMV真核启动子替代原核启动子SP6,其能直接将基因组以DNA形式转染至细胞并开始转录翻译。黄病毒cDNA在大肠杆菌宿主中极不稳定,潜在的原核启动子使基因组产生针对大肠杆菌的毒性蛋白,使其很难在大肠杆菌中稳定传代[21]。针对这样的原核毒性,已经有设计多种策略来应对,包括使用极低拷贝数质粒、多片段系统、体外连接方法、插入内含子以及用于质粒繁殖的酵母作为替代宿主菌[22-28]。原有的单拷贝质粒载体pCC1虽然能极大程度减少原核毒性,但是由于质粒拷贝数过低,使获取足量的质粒用于下游实验成为问题。因此,根据本实验室在构建TMUV复制子以及感染性克隆[8,29]的前期经验,我们选用低拷贝质粒作为载体,既能稳定YFV17D基因组,又能得到一定量的质粒拷贝数完成下游实验。
在构建YFV17D复制子的策略上,我们保留了完整的C蛋白和全部非结构蛋白,删除了prM蛋白和E蛋白的绝大部分。此外,E基因的C端序列保留了34个氨基酸,用于编码下游NS1蛋白的信号序列,信号序列的保留能保证NS1蛋白的正确加工以及非结构蛋白在内质网膜上的正确转运[6,30]。此外,构建表达缺失的结构蛋白(prM和E)的质粒作为包装质粒(pcDNA3.1-CprME和pcDNA3.1-C28prME)以包装目的复制子,其中,pcDNA3.1-C28prME保留C蛋白的C端28个密码子(C28),作为 prM 蛋白易位进入内质网腔的信号序列; pcDNA3.1-CprME表达YFV17D的全部结构蛋白。虽然,我们设计的两种包装质粒都成功打包了复制子基因组,但是两种包装质粒呈现出不同的包装效率。从结果来看,无论是检测Nluc活性,还是检测荧光蛋白的表达,CprME包装效率都明显高于C28prME。Roby等[31]构建的KUNV亚基因复制子删除了KUNV C蛋白第18-100位密码子,保留了完整的prM、E蛋白和非结构蛋白,并在同一质粒中使用两个CMV启动子分别表达KUNV的完整C蛋白和复制子亚基因组。在转染细胞后能够产生低水平的SRIPs。但通过使用EF1α启动子替换驱动C蛋白的CMV启动子时,不仅能表达出更高水平的C蛋白,而且同时还能显著提高SRIPs的产生[32]。这解释了不同包装质粒对YFV17D不同的包装效率。在共转染体系中,在YFV17D复制子基因组表达完整C蛋白的同时,包装质粒CprME比C28prME能额外提供完整C蛋白,使整个体系的C蛋白表达的量增加,更多的C蛋白与prM蛋白能更多地打包复制子基因组,从而提高包装效率。
4 结论
本研究利用反向遗传学技术成功构建携带报告基因的YFV17D复制子,通过与表达YFV17D结构蛋白的真核表达质粒共同转染至BHK-21细胞,成功包装出具有单轮感染能力的假病毒颗粒。
猜你喜欢
今日农业(2021年11期)2021-08-13
食品科学(2018年10期)2018-05-23
中国药理学通报(2015年2期)2016-01-12
热带农业科学(2015年9期)2015-10-14
西南医科大学学报(2015年1期)2015-08-22
中国医学科学院学报(2015年5期)2015-03-01
中国当代医药(2015年9期)2015-03-01
西南军医(2015年6期)2015-01-23
遗传(2014年3期)2014-02-28
世界科学(2014年8期)2014-02-28
