
早期抽薹对当归根际土壤微环境的影响
2023-08-15 08:30谢田朋张佳宁董永骏张建景明
生物技术通报 2023年7期
谢田朋 张佳宁 董永骏 张建 景明
(1.甘肃中医药大学,兰州 730000;2.甘肃祁连山国家级自然保护区管护中心,张掖 734000)
当归[Angelica sinensis(Olive.)Diels]为伞形科多年生草本植物,其干燥根作为补血活血、调经止痛、润肠通便的药物在我国已有上千年的用药历史[1-2]。当归在我国甘肃、贵州、山西、青海、四川、云南等地广泛种植[3],其中,甘肃岷县所产岷归因品质好、产量高素有“岷归甲中华”之美称[4]。当归中含有超过370种化学物质,包括苯酞类、单萜和倍半萜类、芳香类化合物、脂肪烃及其衍生物、多糖及有机酸等[5],其中当归多糖、阿魏酸等化学物质具有抗炎、抗肿瘤、抗抑郁、增强心脑血管功能、免疫调节等多种药理活性[5-7]。
随着当归广谱药效的研究和相关药物的开发,其需求量逐年增加,每年栽培面积超过43 500 hm2[8],但当归在种植中存在的连作障碍[9]、根腐病[10]和早期抽薹[11]等问题仍未得到解决。在目前的种植生产中,当归种子于第一年初夏播种,秋季收集发芽幼苗于室内越冬,第二年春季越冬幼苗继续营养生长,同年秋季采收其非木质化根,或者在田间保存到第三年收获新种[11]。然而,二年生当归中有超过40%的个体发生早期抽薹现象,这会让当归根部木质化并失去药用价值[11-12]。当归早期抽薹是一个复杂的植物生化过程,幼苗年龄、幼苗重量和品种等内源因素,以及温度、光照、施肥和海拔等外部因素均会导致该现象的发生[12-15]。但是,在实际种植过程中发现,在相同的内源和外源因素控制下,部分当归仍会发生早期抽薹现象,这表明当归早期抽薹可能受到一些尚未完全了解的因素影响。
前期研究表明,当归根际土壤微生物群落会随着生长阶段的不同而发生改变,并且抽薹期当归根际土壤中部分微生物的相对丰度与其他生长阶段存在显著变化[16],这说明早期抽薹与根际土壤微生物可能存在联系。根际土壤是植物生长、繁殖和代谢活动的最关键区域,植物根系可以通过分泌各种代谢物来影响其微生物群,有助于驱动根际微生物群落的组成[17]。同时,根际土壤微生物群也可能影响宿主植物的代谢,研究表明,拟南芥(Arabidopsis thaliana)根系分泌物会影响根际微生物的群落组成,而微生物可以通过将色氨酸转化为植物激素吲哚乙酸(IAA)来下调植物中开花有关基因,从而延缓开花,并且微生物还可以通过硝化作用增加和延长氮的生物可利用性,刺激植物进一步营养生长[18]。因此,研究根系微生物及代谢物对植物表型可塑性的影响对增加经济作物产量上具有重要意义[19]。
目前,关于当归早期抽薹现象与根际土壤细菌群落及土壤代谢的相关研究鲜有报道。因此,本研究通过控制试验,利用16S rDNA扩增高通量测序和气相色谱-质谱联用方法(GC-MC),观测抽薹与未抽薹当归根际土壤细菌群落及代谢组变化,并寻找当归根际土壤性质、细菌群落及代谢物之间在当归抽薹现象中的关联性,通过研究当归早期抽薹对根际土壤微环境的影响,尝试为解决当归抽薹问题提供新的思路。
1 材料与方法
1.1 材料
供试当归为‘岷归1号’,购自定西市农业科学研究院。为避免内源因素对当归抽薹的影响,根据文献记载[11],试验中选用百苗重约100 g和直径约0.55 cm的二龄幼苗作为供试材料。由于当归为“低温长日照”型植物[11],为避免光照等外源因素对当归抽薹的影响,试验在遮阴度25%的小型遮阴棚中开展,当归幼苗移栽在盆口直径16 cm、高20 cm的黑色塑料育苗盆中,1株/盆,共100盆。
1.2 方法
1.2.1 试验地概况 试验地位于甘肃中医药大学杏林百草园种植基地(103°95'E,35°97'N),当地海拔1 750 m,年平均气温15.5℃,年平均降雨量900 mm,无霜期290 d。供试土壤类型为黄土,实验前土壤pH 7.77(土水比1∶2.5)、盐分7.53 mg/kg、有机质3.77 g/kg,全氮1.38 g/kg、全磷3.32 g/kg、全钾3.45g/kg、硝态氮39.32 mg/kg、氨态氮20.11 mg/kg、有效磷37.24 mg/kg、有效钾172.47 mg/kg。
1.2.2 试验设计 试验用肥为(NH4)2HPO4复合肥料,3 g/盆,在移栽的同时拌施于土壤中,后期不再追肥。试验过程中,每20 d对花盆进行一次随机位置调整,避免环境差异对个体的影响,并根据天气情况,定期对花盆进行浇灌,用水量为500 mL/盆。3个月后当归生长进入早期抽薹期,观察发现试验中20%的当归发生了早期抽薹现象。
1.2.3 样品采集 当归根际土壤于2021年7月19日采集。随机选取抽薹和未抽薹当归各18盆,用剪刀剪开花盆后取出当归,将根部土壤抖落后用无菌毛刷将附着于根部的土壤轻轻刷落,每3株当归的根际土壤混合在一起后分成两份,一份装于无菌冻存管中并迅速置于干冰中暂时存放,用于土壤细菌和代谢物的检测,另一份放入自封袋内,用于土壤理化性质测定。抽薹和未抽薹组各设置6次重复。
1.2.4 土壤理化指标测定 密封于自封袋中的土壤风干后过60目细筛,采用pH计测定pH值、TDS计测定盐分、有机质采用重铬酸钾氧化-外加热法、全氮采用凯氏定氮法、铵态氮采用氯化钾浸提-靛酚蓝比色法、硝态氮采用紫外分光光度计法、全磷采用酸溶-钼锑抗比色法、有效磷采用钼锑抗比色法、全钾采用氢氧化钠熔融-火焰光度法、有效钾采用乙酸铵浸提-火焰光度法测定[20]。
1.2.5 土壤细菌群落DNA提取及测序 土壤DNA提取采用土壤总DNA提取试剂盒DNeasy PowerSoil Kit(QIAGEN,德国)提取。使用前端343F引物5'-TACGGRAGGCAGCAG-3'和后端798R引物5'-AGGGTATCTAATCCT-3'对16S rDNA基因的V3-V4区进行聚合酶链式反应(polymerase chain reaction,PCR)扩增,每个样品在30 μL体系中扩增,该反应第一轮体系包括15 μL 2×Gflex PCR 缓冲液,0.6 μL Tks Gflex DNA 聚合酶(1.25 U/μL),1 μL模板,5 pmol/μL正反引物各1 μL。在94℃进行5 min初始变性后,将目标区域在94℃进行30 s,56℃进行30 s,72℃进行20 s的26个循环下扩增,最终72℃下进行5 min。用1%琼脂凝胶电泳验证PCR结果,若阴性未出带,则用AMPure XP试剂盒(BECKMAN COULTER,美国)进行磁珠纯化,纯化后稀释至50 μg/μL作为PCR模板进行第二轮扩增,在与第一轮PCR相同条件下将DNA样品扩增7个循环。取5 μL纯化过的二轮产物进行1%琼脂糖凝胶电泳检测,检测是否有条带和条带是否单一,取1 μL纯化过的二轮产物在Nanodrop进行浓度检测。样品质检合格后则交由测序公司利用Illumina HiSeq PE250测序平台进行16S rDNA扩增测序(欧易生物,上海)。
1.2.6 土壤代谢组提取及分析条件
1.2.6.1 土壤代谢物提取 将20 μL内标(L-2-氯-苯丙氨酸,0.06 mg/mL,甲醇配置)和1 mL甲醇-水(V∶V=1∶1)加入500 mg土壤中,样品在-20℃放置2 min预冷后放入组织研磨机中研磨2 min。研磨样品以7 700 r/min低温离心10 min(4℃),冷冻干燥后溶于200 μL甲醇-水(V∶V=1∶1)复溶,溶解后的样品在12 000 r/min低温离心10 min(4℃),取150 μL上清液装入玻璃衍生瓶中。将所有样本的提取液等体积混合制备质控样本。所有样品和质控样本经冷冻干燥后,加入80 μL的甲氧胺盐酸盐吡啶溶液(15 mg/mL),涡旋振荡2 min后,在37℃振荡培养箱中肟化反应90 min。所有样品加入50 μL BSTFA(含1% TMCS)衍生化试剂、20 μL的正己烷、10 μL混合内标溶液(C8/C9/C10/C12/C14/C16,0.16 mg/mL,C18/C20/C22/C24,0.08 mg/mL,均为氯仿配置),混合样品在70℃下反应60 min后进行气象色谱-质谱(GC-MS)代谢组学分析,每个处理进行6个独立生物学重复。
1.2.6.2 GC-MS分析条件 土壤代谢物采用安捷伦7890B气相色谱系统和安捷伦5977A MSD系统耦合分析。色谱条件:采用DB-5MS毛细管柱(30 m×0.25 mm×0.25 μm)分离,载气为高纯氦气,流速为1.0 mL/min。进样口的温度为260℃,进样量1 μL。柱温箱的初始温度为60℃,保持0.5 min,然后以8℃/min的速度增加到210℃,再以15℃/min的速度增加到270℃,最后以20℃/min的速度增加到305℃,并保持5 min。质谱条件:MS四极子和离子源(电子冲击)的温度分别为150℃和230℃,碰撞能量为70 eV,采用全扫描模式(SCAN,m/z50-500)获取大量数据。
1.2.6.3 质谱数据的定性和相对定量 GC-MS定性采用鹿明生物自主研发的LUG数据库(Untargetdatabase of GC-MS from Lumingbio),该数据库中包含2 543种能够采用GC-MS检测的代谢物,其质荷比(m/z)范围为85-650。对样本的所有峰值信号强度(峰值面积)进行筛选并分段归一化(阈值:内标RSD<0.3),对归一化数据进行去冗余和峰合并,得到最终代谢产物。
1.2.7 数据分析
1.2.7.1 细菌群落测序数据分析 原始测序序列为FASTQ格式,使用Trimmomatic软件进行去杂质控,过滤reads尾部质量值20以下的碱基。去杂后的双端序列使用FLASH软件进行拼接,将成对reads拼接成1条序列,同时利用UCHIME检测并去除序列中的嵌合体序列。采用Vsearch 软件,将序列相似性大于或等于97%的归为一个OTU单元,最终得到多个 OTU。使用 QIIME软件包挑选出各个OTU的代表序列,并使用RDP classifier软件将所有代表序列与Greengenes或者Silva(version123)数据库进行比对注释,并保留置信区间大于0.7的注释结果。
Alpha多样性分析采用QIIME和R包vegan 2.5-6 实现,主要进行了Chao1指数、Shannon指数、Simpson指数计算,主成分分析(PCA)采用https://cloud.oebiotech.cn/task/平台运算绘图,采用SPSS20.0软件(SPSS Inc., Chicago, IL)对不同微生物类群丰度进行显著性差异分析,采用方差分析(ANOVA)和最小显著性差异(LSD)进行事后比较检验(显著性水平P<0.05),柱状图在Origin2022中绘制。线性判别分析[linear discriminant analysis(LDA)effect size(LefSe)]采用https://www.omicstu dio.cn/tool/60平台运算绘图,将LDA值>3.5作为丰度显著差异的判定值。
1.2.7.2 根际土壤代谢物数据分析 采用SPSS20.0软件(SPSS Inc., Chicago, IL)对代谢物进行显著性差异分析,采用方差分析(ANOVA)和最小显著性差异(LSD)进行事后比较检验(显著性水平P<0.05)。PCA、差异代谢物火山图、组间差异热图及通路富集气泡图用https://www.metaboanalyst.ca/平台分析绘制。
1.2.7.3 土壤性质、差异微生物及差异代谢组互作分析 采用冗余分析/典范应对分析(RDA/CCA)分析土壤性质、差异微生物、差异代谢物之间的两两关系。RDA/CCA分析采用https://www.omicshare.com/index.php平台分析进行。采用Pearson相关性分析对筛选出的土壤元素、差异微生物与富集在关键通路上的差异代谢物进行关联分析。
2 结果
2.1 根际土壤理化性质
硝态氮(P=0.040)含量在抽薹当归根际土壤中显著升高,而其他理化指标均在抽薹和未抽薹当归根际土壤间无显著差异(P>0.05)(表1)。
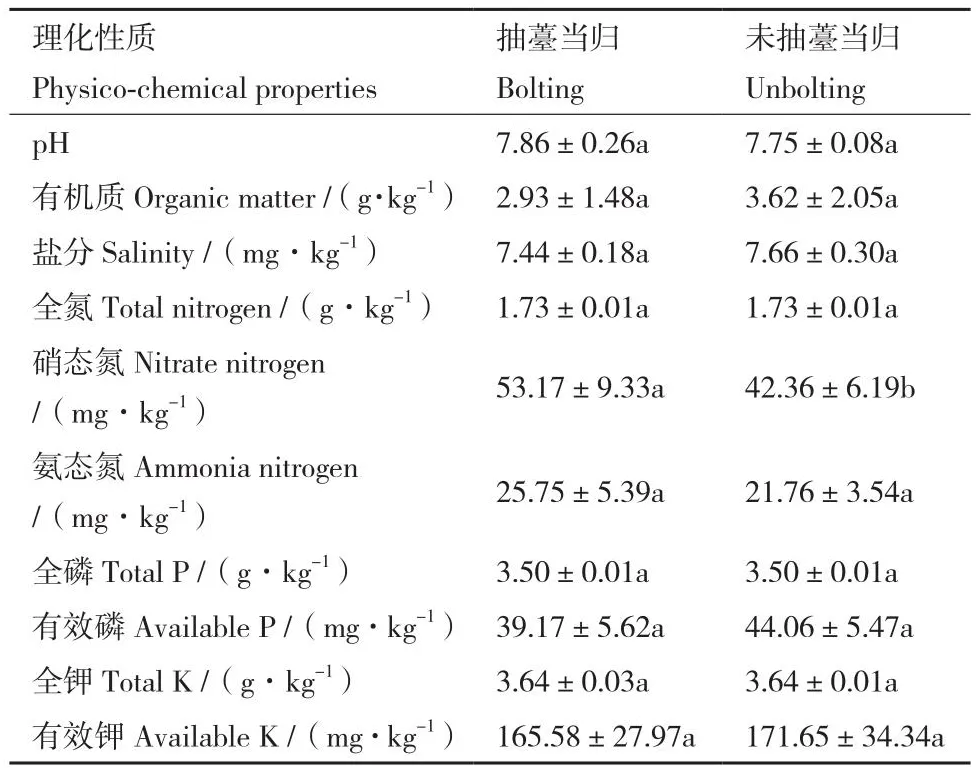
表1 抽薹与未抽薹当归根际土壤的理化性质Table 1 Physico-chemical properties of the rhizosphere soils of bolting and unbolting Angelica sinensis
2.2 根际土壤细菌群落结构
本试验在抽薹和未抽薹当归根际土壤中共检测到5 554个细菌OTUs。与根际土壤理化性质结果相似,所有 Alpha多样性指数均在抽薹和未抽薹当归根际土壤间无显著差异(P>0.05)(表2)。如主成分分析(PCA)所示(图1),其中PC1解释了变量的60.1%,PC2解释了变量的25.88%,可以看到,抽薹与未抽薹当归根际土壤细菌群落不能沿横纵坐标轴发生明显分离。结果表明,抽薹和未抽薹当归根际土壤细菌群落并没有发生明显的丰富度和均匀度变化,群落结构是相似的。

图1 抽薹(BO)与未抽薹(UB)当归根际土壤细菌群落主成分分析Fig.1 Principal component analysis plots of the bacterial communities in the rhizosphere soils of bolting(BO)and unbolting(UB)A.sinensis

表2 抽薹与未抽薹当归根际土壤细菌群落的阿尔法多样性Table 2 The alpha diversity of bacterial communities in rhizosphere soil of bolting and unbolting A.sinensis
抽薹和未抽薹当归根际土壤细菌群落在门水平和属水平上的优势菌群组成一致,分析表明,菌群相对丰度在组间无显著差异(P> 0.05)(表3)。在相对丰度前15的细菌门中,变形菌门(Proteobacteria)、放线菌门(Actinobacteriota)、拟杆菌门(Bacteroidota)、芽单胞菌门(Gemmatimonadota)、厚壁菌门(Firmicutes)、黏球菌门(Myxococcota)的平均占比达97%以上,是当归根际土壤中最主要的优势菌门。当归根际土壤中相对丰度前15的优势细菌属为鞘氨醇单胞菌属(Sphingomonas)、链霉菌属(Streptomyces)、假黄单胞菌属(Pseudoxanthomonas)、原小单孢菌属(Promicromonospora)、MND1、溶杆菌属(Lysobacter)、Allorhizobium-Neorhizobium-Pararhizobium-Rhizobium属、Muribaculaceae属、Nocardioides属、Ellin6067、大肠杆菌志贺菌属(Escherichia-Shigella)、纤维弧菌属(Cellvibrio)、假单胞菌属(Pseudomonas)、Sphingopyxis、0319-7L14,占总相对丰度的27%以上(表3)。这些结果进一步说明,抽薹与未抽薹当归根际土壤优势菌群的结构是相似的。
线性判别分析(LefSe)结果(图2)显示,将LDA 值>3.5 定义为微生物在数量上差异显著,这部分微生物叫作响应微生物(responder),代表这部分微生物数量对抽薹现象响应显著。由图2可知,有3个科和10个属的非优势菌群在抽薹和未抽薹当归根际土壤中存在明显的差异。在抽薹当归根际土壤中,伦黑墨氏菌属(Rheinheimera)的相对丰度显著高于未抽薹当归(P= 0.037);而珊瑚状放线菌属(Actinocorallia)(P= 0.025)、肠杆菌属(Enterorhabdus)(P= 0.024)、Phaselicystis属(P= 0.010)、柄杆菌属(Caulobacter)(P= 0.010)、Agaricicola属(P= 0.007)、Rubellimicrobium属(P= 0.010)、纤毛菌属(Leptothrix)(P= 0.025)、亚硝酸菌属(Nitrosomonas)(P= 0.025)、不动杆菌属(Acinetobacter)(P= 0.010)的相对丰度则显著低于未抽薹当归,其中,Agaricicola属和亚硝酸菌属在抽薹当归根际土壤中几乎或完全消失。
2.3 根际土壤代谢物变化
采用气相色谱-质谱联用方法(GC-MS)在所有土壤样本中共检测并鉴定到了包括有机酸、有机氧化合物、脂类、苯类、有机杂环化合物等301个代谢物。在这些化合物中,有机酸和有机氧化合物的数量最多,分别占代谢物总数的20.2%和20.2%,其次是脂类(14.9%)、苯类(8.9%)、有机杂环化合物(7.9%)(图3-A)。PCA分析表明,抽薹当归和未抽薹当归根际土壤的代谢物存在明显差异,并沿着纵坐标轴分离(图3-B)。
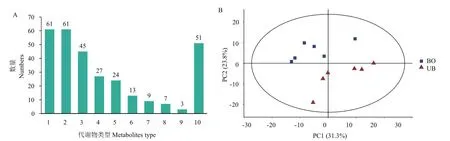
图3 抽薹(BO)与未抽薹(UB)当归根际土壤代谢物类型(A)和主成分分析(B)Fig.3 Metabolite type distribution(A)and the principal component analysis plots(B)of the rhizosphere soils metabolites of bolting(BO)and unbolting(UB)A.sinensis
301种代谢物中,有66种的含量在组间差异显著(P<0.05, VIP>1),抽薹当归组中有52种代谢物显著上调,14种代谢物显著下调(图4-A),上调的代谢产物主要属于有机酸及其衍生物、苯类化合物,下调的代谢产物主要属于脂质和类脂分子(图4-B)。其中,上调最多的5种代谢物是N-甲基-L-丙氨酸(N-methy-L-alanine)、松三糖(melezitose)、氢化肉桂酸(hydrocinnamic acid)、L-半胱氨酸甘氨酸(L-cysteine-glycin)、N-氨基甲酰谷氨酸(N-carbamylglutamate);下调最明显的是红四氟呋喃糖(erythrotetrofuranose)、四糖酸(tetracosanoci acid)、丁内酰胺(butyrolactam)、莽草酸(shikimic acid)、甲基β-d-吡喃葡萄糖苷(methyl beta-dglucopyranoside)(图4-C)。通路富集分析可用以阐明根际土壤代谢过程的具体变化,富集通路分析(图4-D)表明,抽薹当归组有13条通路在P<0.05水平上发生改变,其中7条在P<0.01水平上发生改变。这7条通路主要涉及氨基酸代谢、异种生物降解和代谢、脂质代谢,其中氨基酸代谢包括苯丙氨酸代谢(phenylalanine metabolism; ko00360)、苯丙氨酸、酪氨酸和色氨酸生物合成(phenylalanine,tyrosine and tryptophan biosynthesis; ko00400)、酪氨酸代谢(tyrosine metabolism; ko00350)、精氨酸和脯氨酸代谢(arginine and proline metabolism; ko00330)4条通路;异种生物降解和代谢包括氨基苯甲酸酯降解(aminobenzoate degradation ko00627)和苯乙烯降解(styrene degradation; ko00643)2条通路;脂质代谢包括不饱和脂肪酸的生物合成(biosynthesis of unsaturated fatty acids; ko01040)通路。

图4 抽薹(BO)vs未抽薹(UB)当归根际土壤代谢物变化Fig.4 Changes of metabolites in the rhizosphere soils between bolting(BO)and unbolting(UB)A.sinensis
2.4 根际土壤理化性质、差异细菌属、差异代谢物的相关性分析
为揭示土壤理化性质、差异细菌属及差异代谢物之间的相关性,对三者进行两两RDA/CCA分析(冗余分析/典范应对分析)。首先进行除趋势对应分析(detrended correspondence analysis, DCA),结果显示4个轴中的特征值最大均小于3.0,故选择线性RDA模型进行后续分析。在土壤理化指标与差异细菌属的RDA模型中(图5-A),RDA1和RDA2轴特征值的贡献率分别为40.31%和25.68%,总贡献率为65.99%。分析表明,10个土壤指标对抽薹与未抽薹当归的差异细菌属构成均无显著影响(P>0.05),其中,硝态氮的P值为0.061,是所有指标的P值最小值,它与抽薹当归差异细菌属存在正相关趋势,与未抽薹当归存在负相关趋势。
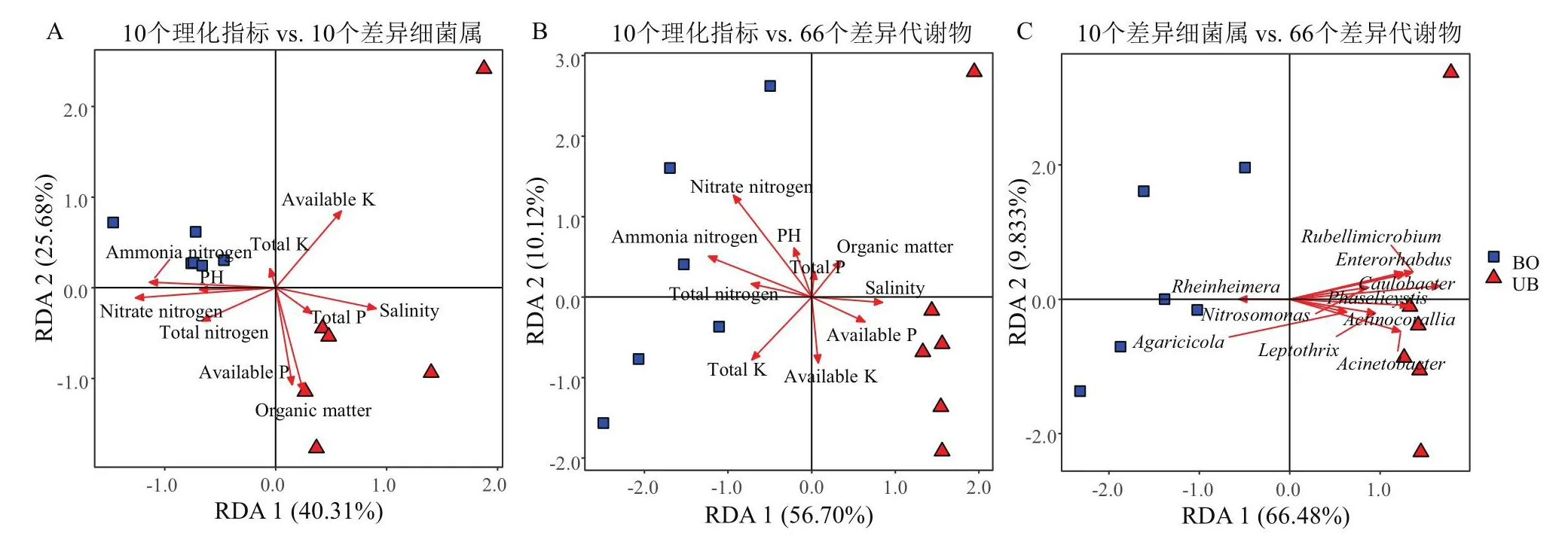
图5 抽薹(BO)与未抽薹(UB)当归根际土壤理化指标与差异细菌属(A)、土壤理化性质与差异代谢物(B)、差异细菌属与差异代谢物(C)的冗余分析Fig.5 RDA analysis of soil properties and different bacterial genus(A), soil properties and different metabolites(B),different bacterial genus and metabolites(C)between bolting(BO)and unbolting(UB)A.sinensis
在土壤理化指标与差异代谢物的RDA模型中(图5-B),RDA1和RDA2轴特征值的贡献率分别为56.70%和10.12%,总贡献率为66.82%。结果表明,只有硝态氮(P=0.049)对抽薹与未抽薹当归的差异代谢物构成有显著影响,如图所示,硝态氮与抽薹当归差异代谢物含量正相关,与未抽薹当归负相关。
在土壤差异细菌属与差异代谢物的RDA模型中(图5-C),RDA1和RDA2轴特征值的贡献率分别为66.48%和9.83%,总贡献率为76.31%。结果表明,Phaselicystis属(P=0.007)、Rubellimicrobium属(P=0.036)对抽薹与未抽薹当归的差异代谢物构成有显著影响,如图所示,二者均与未抽薹当归差异代谢物含量变化正相关,与抽薹当归负相关。
为进一步揭示因当归抽薹引起的7条主要代谢通路中的差异代谢物与硝态氮、Phaselicystis属、Rubellimicrobium属的关系,进行了Pearson相关性分析,结果(表4)表明,Phaselicystis属与氨基苯甲酸酯降解、精氨酸和脯氨酸代谢通路中的差异代谢物普遍存在显著的负相关性(P<0.05),Rubellimicrobium属与精氨酸和脯氨酸代谢通路中的差异代谢物存在显著的负相关性(P<0.05)。结合代谢物在各个通路上表达量的变化可以得知,抽薹当归根际土壤中Phaselicystis属、Rubellimicrobium属相对丰度的下降与氨基苯甲酸酯降解、精氨酸和脯氨酸代谢通路上差异代谢物的上调有较大的关联。而硝态氮含量则与不饱和脂肪酸的生物合成通路中的差异代谢物密切负相关。
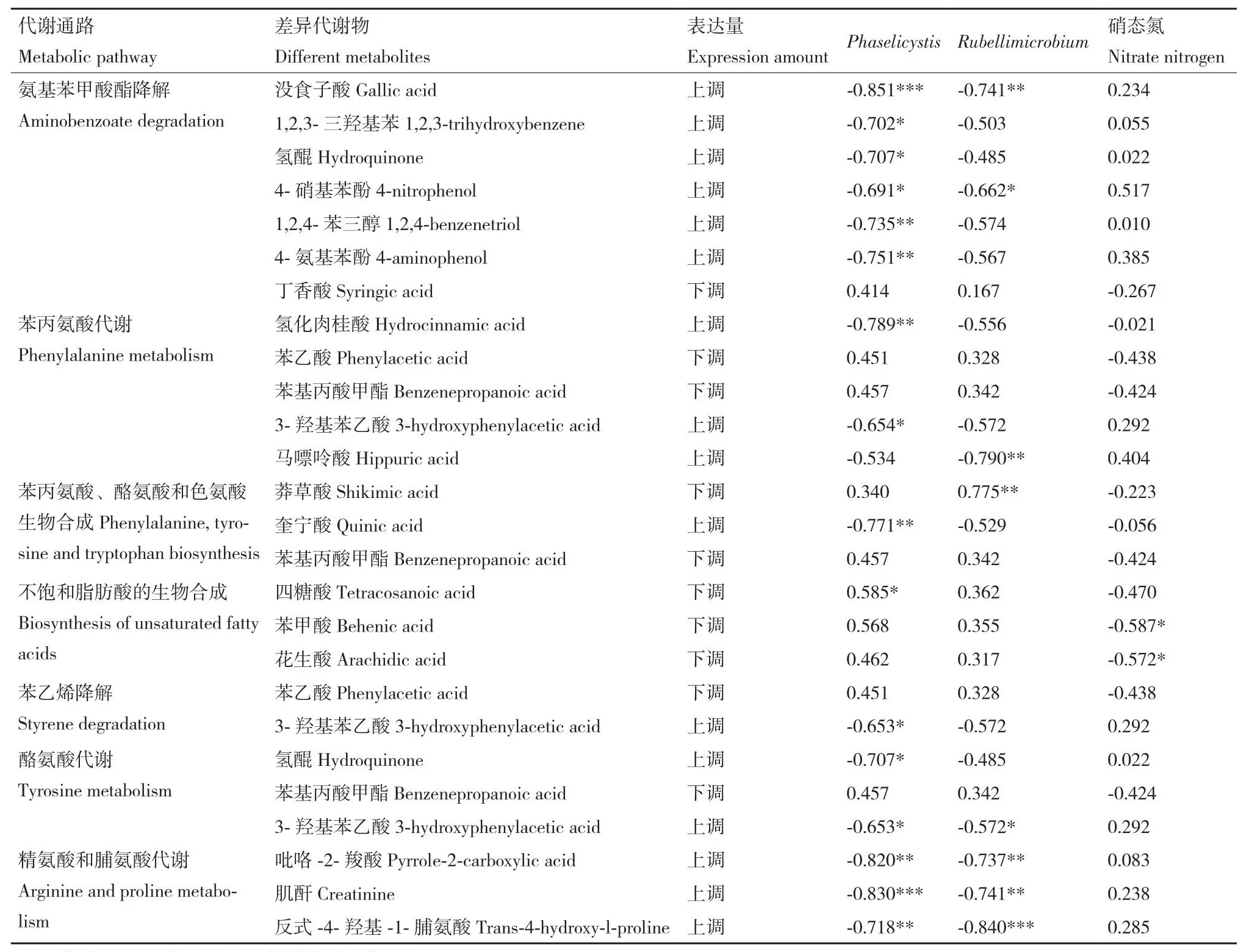
表4 主要代谢通路中差异代谢物与Phaselicystis、Rubellimicrobium、硝态氮含量的相关性Table 4 Correlation between differential metabolites in main metabolic pathways and Phaselicystis, Rubellimicrobium and nitrate nitrogen
3 讨论
本实验主要研究了抽薹与未抽薹当归根际土壤性质、细菌群落和代谢物的变化。研究结果证明,抽薹与未抽薹当归的根际土壤中硝态氮含量、非优势细菌属和代谢物存在显著差异,且彼此间存在一定的联系。这一发现对进一步理解当归抽薹现象的发生尤为重要。
3.1 抽薹当归根际土壤中硝态氮含量上升
本研究发现,硝态氮含量在抽薹当归根际土壤中显著增加,这一现象的发生有两种可能。一方面,当归在早期抽薹期间减少了对硝态氮的吸收,从而增加了根际土壤中硝态氮的含量,这可能与植物在特定生长时期的营养偏好有关[21]。另一方面,硝态氮含量升高能够促进当归早期抽薹的发生,这是因为氮素是植物开花和花序发育所必需的营养元素。有研究表明,在氮总量不变的情况下,植物成花率会随着NO3-/NH4+的增大而升高[22]。硝态氮不仅是植物生长发育所必需的重要营养元素,也是重要的信号分子[23],例如在有关茶树的研究中发现,一种抑制开花的基因miR156在NO3-含量增加后被抑制,从而起到促进开花的作用[24]。当归早期抽薹与根际土壤硝态氮含量升高间的因果关系还需在今后的研究中进一步明确。
3.2 根际土壤非优势细菌属相对丰度显著变化
抽薹与未抽薹当归根际土壤细菌群落结构和组成是相似的,其优势细菌门和优势细菌属组成与前期研究结果基本一致[16]。可见,当归根际土壤细菌群落并不会随植物的生长周期发生显著变化,菌群维持平衡状态对植株保持健康生长可能具有重要意义。虽然当归根际土壤细菌群落不会发生较大的变化,但一些非优势菌属却在组间发生改变,目前已有研究证实非优势菌属在影响植物生长发育中发挥着作用[25]。虽然RDA分析中硝态氮含量对差异细菌属相对丰度影响并不显著,但其与抽薹和未抽薹当归根际土壤中差异细菌的相关性仍是所有土壤性质指标中最明显的,这说明硝态氮仍是非优势菌属丰度变化中最值得考虑的因素。土壤的NH4+可以通过硝化细菌的硝化作用转变为NO3-,进而参与植物的生长发育过程,而NO3-亦会被反硝化细菌还原为N2[26]。目前没有证据证明在抽薹当归根际土壤中丰度上升的Rheinheimera属为硝化细菌,同时丰度下降的9个细菌属中,只有Nitrosomonas属被证明是反硝化细菌[27],而在抽薹当归根际土壤中,Nitrosomonas属完全消失。因此,猜测抽薹当归根际土壤中硝态氮含量的增加可能与Nitrosomonas属的缺失有关。
3.3 根际土壤代谢物含量显著变化
本研究表明,当归抽薹可引起根际土壤代谢物的明显变化。植物根际土壤中的代谢物来源于植物根系分泌、微生物代谢及植物、微生物和土壤有机质的分解[28],本研究发现,抽薹与未抽薹当归根际土壤中的有机质含量及细菌群落多样性和优势菌群并未产生显著差异,因此,根际土壤中的差异代谢物主要源于根系分泌物的可能性较大。Song等[29]通过对辣椒的研究也发现根际土壤代谢物的主要来源为根系分泌物。代谢物作为直接反应植物生理状态的物质,直观地反应在植物的表型特征上。研究表明,根系分泌物可引起土壤菌群组成及功能的变化[30],本研究中,当归抽薹过程使氨基酸代谢、异种生物降解和代谢及脂质代谢的相关通路上发生代谢物质含量变化,这些代谢物通过根系分泌到土壤中后引起根际土壤非优势菌群的变化,其中Phaselicystis属和Rubellimicrobium属与代谢物的变化密切相关(P<0.05),且主要与氨基苯甲酸酯降解、精氨酸和脯氨酸代谢通路中差异代谢物显著负相关。据报道,Phaselicystis属具有溶菌和非纤维素分解能力,还能产生多不饱和脂肪酸ω-6花生四烯酸[31],而Rubellimicrobium属则具有降解大分子有机物的能力[32],但目前并不清楚Phaselicystis属和Rubellimicrobium属在当归抽薹中是否发挥具体作用。尽管硝态氮含量与抽薹当归差异代谢物间有显著的关联性(P<0.05),但与Phaselicystis属和Rubellimicrobium属不同,其主要与不饱和脂肪酸的生物合成通路上的差异代谢物显著负相关。由此可见,虽然菌属和营养元素都会与差异代谢物产生负相关性,但作用的代谢通路并不相同,其相关机制也必然不相同。
3.4 当归抽薹与硝态氮、非优势细菌和代谢物变化的关联
当归根际土壤NO3-含量上升与当归早期抽薹现象存在联系,但彼此间的因果关系仍需进一步研究验证。当归抽薹后引起体内代谢水平变化,并以根际分泌物的形式进入根际土壤,进而导致根际土壤非优势菌群的丰度变化,其中Phaselicystis属和Rubellimicrobium属的丰度下降与氨基苯甲酸酯降解、精氨酸和脯氨酸代谢通路中差异代谢物上调的联系最为紧密,但并无证据表明其在当归早期抽薹中发挥可能的作用。而根际土壤中NO3-含量上升与Nitrosomonas属的变化可能存在联系,但仍需进一步研究论证(图6)。

图6 当归早期抽薹对根际土壤微环境的影响Fig.6 Effects of premature bolting on the rhizosphere soil microenvironment of A.sinensis
4 结论
本文对抽薹与未抽薹当归根际土壤进行了土壤理化性质、细菌群落和代谢物变化研究。二者在根际土壤硝态氮含量、代谢物含量和非优势菌群的相对丰度间均存在显著差异。本研究从当归早期抽薹引起的根际土壤微环境变化角度为切入点,尝试为解决当归早期抽薹问题提供新的思路。
猜你喜欢
现代临床医学(2022年4期)2022-09-29
中国土壤与肥料(2021年5期)2021-12-02
天然产物研究与开发(2018年3期)2018-05-07
中国蔬菜(2016年8期)2017-01-15
环境污染与防治(2016年12期)2016-03-13
分析测试学报(2015年7期)2016-01-13
质谱学报(2015年5期)2015-03-01
食品科学(2013年15期)2013-03-11
植物营养与肥料学报(2011年4期)2011-10-26
植物营养与肥料学报(2010年3期)2010-11-16
