
基于Super-GBS的高粱株高和节间数QTL定位
2023-08-15 08:30徐建霞丁延庆冯周曹宁程斌高旭邹桂花张立异
生物技术通报 2023年7期
徐建霞 丁延庆 冯周,2 曹宁 程斌 高旭 邹桂花 张立异
(1.贵州省农业科学院旱粮研究所,贵阳 550006;2.贵州大学农学院,贵阳 550025;3.浙江省农业科学院作物与核技术利用研究所,杭州310021)
高粱(Sorghum bicolorL.Moench)作为世界第五大粮食作物,广泛分布于世界的热带干旱和半干旱地区,为全球约5亿人口提供粮食[1]。高粱作为C4作物,具有光合能力强、高生物学产量和高抗逆性(抗涝、抗旱、耐盐碱和耐贫瘠)及基因组相对较小等特点,已逐渐成为禾本科作物基因组研究的一个模式作物[2]。株型是高粱育种的重要组成部分,适宜的株高有益于养分及光合的有效利用、种植密度、抗倒伏,进而有利于提高产量[3]。而且随着我国城镇化推进,农村人口老龄化日益严重,农村发展规模化种植已呈必然趋势,对适合机械化生产的高粱新品种需求不断增加。因此,挖掘控制高粱株高相关QTL,开展遗传机理研究,对利用分子标记辅助育种开展高产、易机化高粱新品种的选育提供理论依据。
目前,用于高粱数量性状基因座(quantitative trait locus, QTL)定位的分子标记技术有SSR(simple sequence repeats)和SNP(single nucleotide polymorphism)等[4-5]。Reddy等[6]利用SSR标记,对籽粒高粱M35-1和B35构建的245份重组自交系群体(recombinant inbred lines, RIL)进行基因型扫描,构建含有237个标记的高粱遗传图谱,确定了10个与株高相关的QTL;苏舒[7]利用83个SSR标记对218个家系的RIL群体(T70/P603)开展基因分型,鉴定了8个高粱株高QTL位点和4个节间数QTL位点;另有研究者利用饲草高粱构建的126个家系RIL群体(不育系Tx623A/苏丹草恢复系Sa)为研究对象,定位到4个控制株高的QTL,对表型的贡献率为24.75%-33.59%[8]。近年来,随着高通量测序技术的发展,基于SNP标记的高密度遗传图谱构建以及重要农艺性状的QTL定位越来越多被报道,这些遗传图谱的标记数量不断增加,标记间的遗传距离减小,连锁群趋于完整,连锁图对全基因组的覆盖率增加,更有利于QTL/基因的精细定位。Zou等[9]利用全基因组重测序技术,构建了包含244个RILs(籽粒高粱654/甜高粱LTR108)的SNP高密度遗传图谱,定位了57个影响高粱抽穗期、株高、节间数等性状的QTL。通过简化基因组测序技术(GBS)对460个RILs(籽粒高粱BTx623/籽粒高粱NOG)的基因分型,Kajiya-kanegae等[10]构建了包含有3 710个SNP标记的高粱遗传图谱,并定位了3个与株高相关性状的QTL。Takanashi等[11]同样利用GBS技术对272个甜高粱RILs(甜高粱Brandes/甜高粱Wray)的基因分型,挖掘到33个影响开花时间、株高和生物量等性状的QTL。目前,在控制株高的4个主要位点中(Dw1-Dw4)[12-13],前3个已经被克隆。Dw1(Sobic.009G230800)编码参与细胞增殖调控的高度保守蛋白[14],Dw2(Sobic.006G067700)编码蛋白激酶[15],而Dw3(Sobic.007G163800)编码ABCB1生长素转运蛋白[16],这些蛋白通过调节茎间长度影响高粱株高。
由于国内外用于QTL定位的高粱遗传图谱主要是“甜高粱/甜高粱”,“甜高粱/籽粒高粱”或“饲草高粱”杂交构建的RILs,对籽粒高粱标记辅助育种的可利用价值具有一定的局限性。
本研究利用籽粒高粱BTx623和贵州茅台酒专用高粱红缨子杂交构建的RIL群体为研究材料,基于简化基因组测序技术构建高粱的高密度遗传连锁图谱,开展株高和节间数相关农艺性状的QTL定位,并鉴定QTL区间内的候选基因,为后续的基因克隆提供依据。
1 材料与方法
1.1 材料
以美国籽粒高粱品种BTx623为母本,贵州茅台酒专用高粱品种红缨子为父本,杂交得到F1代,经单粒传法,构建包含205个家系的F2:7代RIL群体。亲本之间在株型上存在显著差异。BTx623株高较低(1.2-1.5 m),株型紧凑,而红缨子株高较高(2.5-3.0 m),株型松散。亲本与RIL群体分别于2020年在贵阳(2020GY)、安顺(2020AS)和乐东(2020LD),以及2021年在贵阳(2021GY)和安顺(2021AS)5个环境下进行田间种植。采用随机区组设计,3次重复,行距60 cm,行长2 m,株距25 cm,人工点播,适时进行间苗定苗、追肥、除草和病虫害防治等田间管理工作。
1.2 方法
1.2.1 表型调查及数据分析 在高粱成熟期,每个株系随机选择5个单株调查株高(plant height, PH)和节间数(internode numbers, IN),调查方法参照《高粱种质资源描述规范和数据标准》中的相关规定[17]。利用Microsoft excel 2021和SPSS 21.0软件对表型数据的平均值、标准差、变异系数、正态分布检验和相关性进行分析。多环境联合方差分析参照曹永策等[18]方法,根据下面公式计算广义遗传率(h2B)。
式中,根2g为基因型方差,公2ge为基因型与环境互作方差,义2e为环境方差,n为环境数目,r为试验重复数。
1.2.2 DNA提取和GBS重测序 2020年5月在贵阳采集亲本和RIL群体植株嫩叶,采用CTAB法[19]提取DNA,0.8%琼脂糖凝胶检测DNA纯度和完整性,同时用Qubit® 2.0荧光测定计检测DNA浓度。参照Qi等[20]方法,利用上海欧易生物医学科技有限公司建立的GBS技术对质检合格的每个单株DNA样品基因型进行鉴定。
1.2.3 QTL定位 基于已经构建的高密度连锁遗传图谱(包含有1 910个SNP标记,标记间平均遗传间距为0.47 cM)[21],利用QTL IciMapping4.2软件中完备区间作图法(ICIM)[22]对株高和节间数在5个环境下进行QTL检测,步长设定为0.1 cM,随机抽样1 000次,QTL定位的阈值LOD设置为2.5,并计算出每个QTL的贡献率和加性效应,采用“q+性状名称英文缩写+染色体+“.”+QTL序号”的方式对QTL进行命名。
1.2.4 QTL区间内候选基因挖掘方法 在Sorghum QTL网站(http://aussorgm.org.au/sorghum-qtl-atlas/)查找已报道的QTL信息,结合BTx623参考基因组信息,确定候选区间的所有基因,利用植物基因组数据库Phytozome中的高粱参考基因组功能注释信息(https://phytozome-next.jgi.doe.gov/),综合分析Gene Ontology数据库(http://geneontology.org/)和KEGG数据库(https://www.kegg.jp/)预测基因执行的生物学功能,对比水稻中已克隆基因的功能注释来进一步确定候选基因。
2 结果
2.1 农艺性状表型分析和相关性分析
对亲本BTx623、红缨子以及RIL群体在5个环境下株高和节间数表型数据调查结果可知(表1),2个性状在亲本之间差异显著,红缨子的株高和节间数显著高于BTx623。RIL群体2个性状平均值介于双亲之间,且均出现双向超亲现象,5个环境的变异系数介于12.02%-27.46%。此外,RIL群体的2个性状在5个环境下的偏度和峰度绝对值均小于1,除2020LD的节间数峰度为1.6外,且数据分布表现为连续的近似正态分布,符合数量性状遗传特点,适合进行QTL定位分析(图1)。
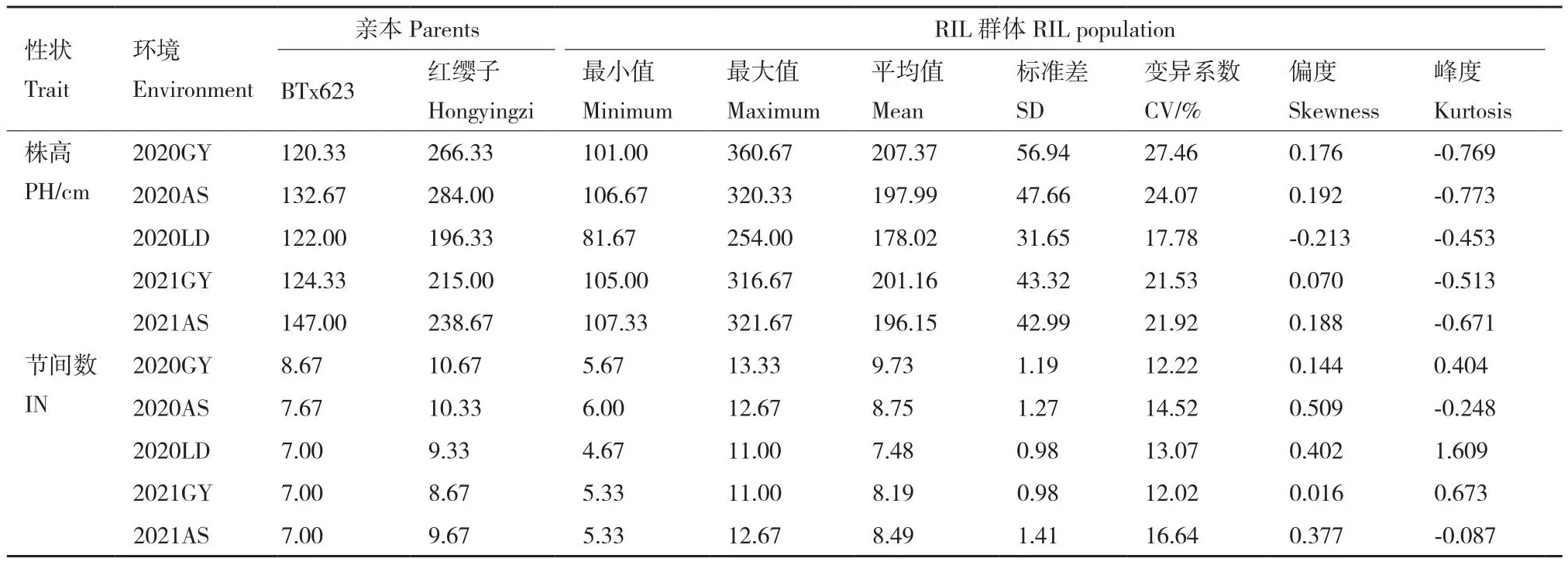
表1 RIL群体中株高和节间数在5个环境下的表型统计Table 1 Phenotypic statistics of plant height and internode numbers in the RIL population in five environments
为了解不同环境下性状间的相关性,对2个性状开展了Pearson相关性分析(表2)。相同性状在不同环境间呈极显著相关性(P<0.01),株高和节间数的平均相关系数R值分别达到0.83和0.68。株高与节间数在不同环境之间也呈显著(P<0.01)正相关关系,R值的范围为0.302-0.884,平均值为0.561。方差分析结果(表3)得出2个性状的基因型、环境及基因型×环境均存在极显著差异(P<0.01),株高和节间数广义遗传率分别为95.23%和85.78%,说明这两个农艺性状的表型差异主要由其本身的遗传因素所决定,而受环境的影响较小。
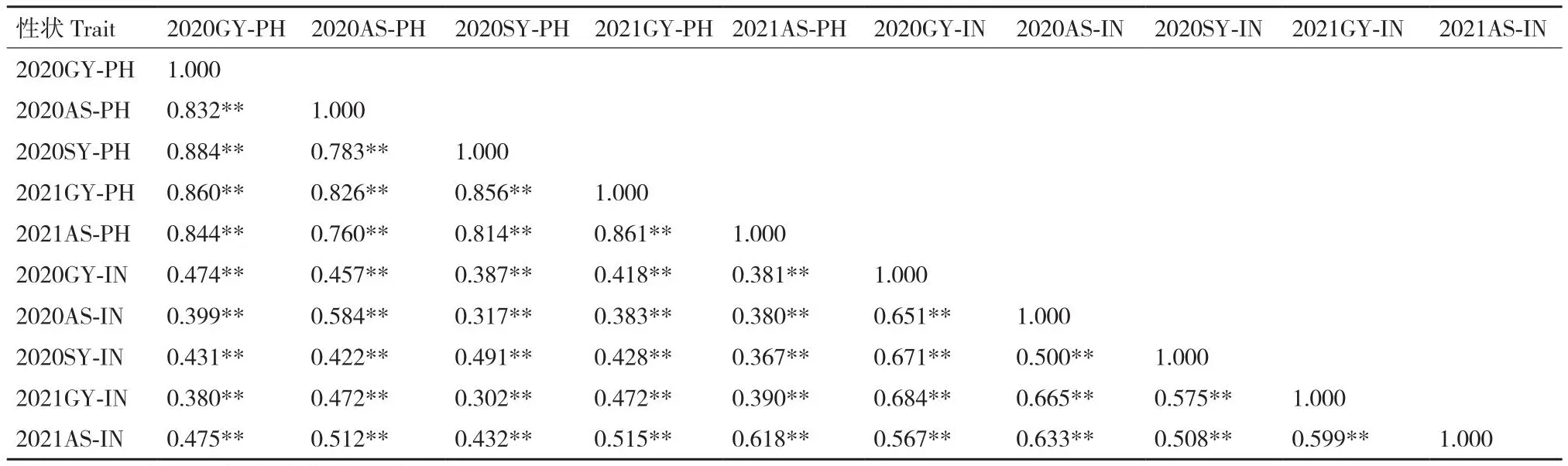
表2 RIL群体中株高和节间数间的相关性分析Table 2 Correlation analysis of plant heighst and internode numbers in the RIL population

表3 株高和节间数间方差分析及广义遗传率Table 3 Variance analysis and broad heritability of plant height and internode numbers
2.2 株高和节间数相关QTL定位
利用ICIM方法,在5个环境下共定位到18个QTL与高粱的株高和节间数相关,分别位于高粱的5条染色体上。与株高和节间数相关QTL分别有7和11个,而2个及以上环境中重复检测到的QTL有10个,涉及到9个染色体区段(表4和图2)。
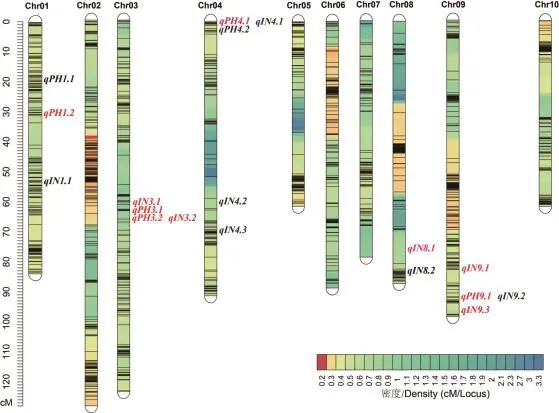
图2 高密度遗传图谱及重要QTL所在染色体示意图Fig.2 Schematic diagram of high-density genetic map and chromosomes with important QTL regions
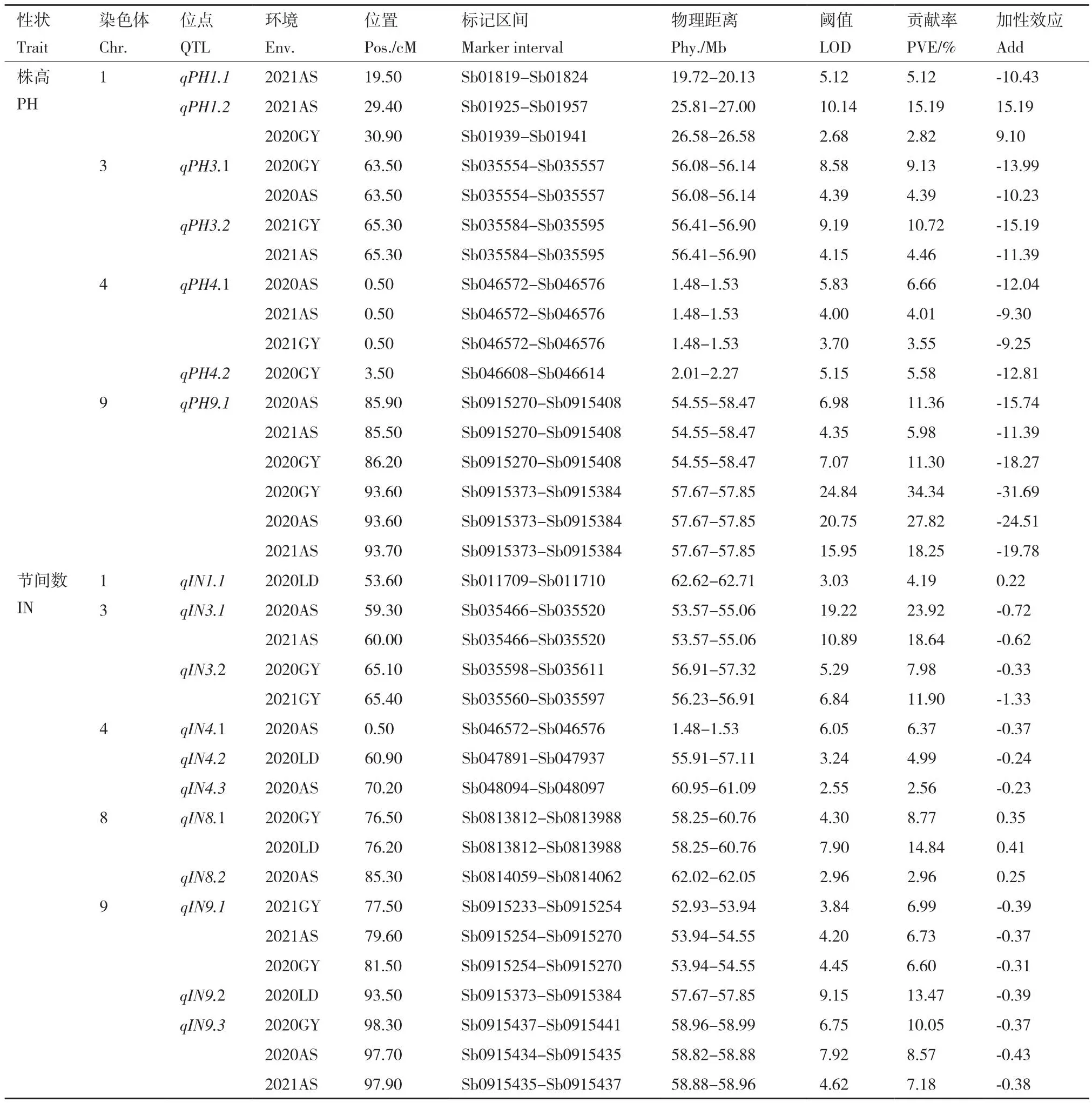
表4 株高和节间数相关QTL定位结果Table 4 QTL mapping results of plant height and internode numbers
影响株高的7个QTL分别位于1、3、4和9染色体上。位于第4和9染色体上的2个重要QTL(qPH4.1和qPH9.1)能够在3个环境下被检测到,最大LOD值分别为5.83和24.84,可解释的最大表型变异率分别为6.66%和34.34%。位于第1染色体的qPH1.2、位于第3染色体qPH3.1和qPH3.2在2个环境下被重复检测到,其最大LOD值和表型贡献率分别为10.14和15.19%。除qPH1.2外,其余6个QTL的增效等位基因均来源于亲本红缨子。
影响节间数的11个QTL分别位于第1、3、4、8和9染色体上。位于第9染色体上的2个重要QTL(qIN9.1和qIN9.3)在3个环境下都能被检测到,它们的最大LOD值分别为4.45和7.92,可解释的最大表型变异率分别为6.99%和10.05%。在11个与节间数相关的QTL中,3个QTL(qIN1.1、qIN8.1和qIN8.2)的增效等位基因来源于亲本BTx623,其余8个QTL的增效等位基因均来源于亲本红缨子。
2.3 重要QTL基因注释和候选基因功能预测
根据上述结果,对2个及以上环境都能定位到的10个重要QTL区段进行了候选基因分析,利用植物基因组数据库Phytozome中的高粱参考基因组功能注释信息(https://phytozome-next.jgi.doe.gov/),识别高粱基因ID、蛋白质注释信息预测基因执行的生物学功能,对比水稻中已克隆基因的功能注释并结合前人研究文献,确定了5个候选基因(表5)。除了已经克隆的控制株高Dw1,还注释到4个新的候选基因。其中,2个基因与水稻株高相关基因同源,包括编码对映型柯巴基焦磷酸合酶的基因Sobic001G248600和编码WRKY转录因子的Sobic003G227300,其水稻同源基因分别为OsCPS1(LOC_Os02g17780)和Dlf1(LOC_Os01g43650)。Sobic008G173300位于控制高粱节间数QTL区间内,蛋白注释为节间伸长抑制因子,其水稻同源基因DEC1(LOC_Os12g42250)编码一种锌指转录因子。Sobic003G227000所在QTL同时控制高粱株高和节间数,该基因编码卵形家族蛋白,在水稻中的同源基因OsOFP2(LOC_Os01g43610)与多个发育过程相关。
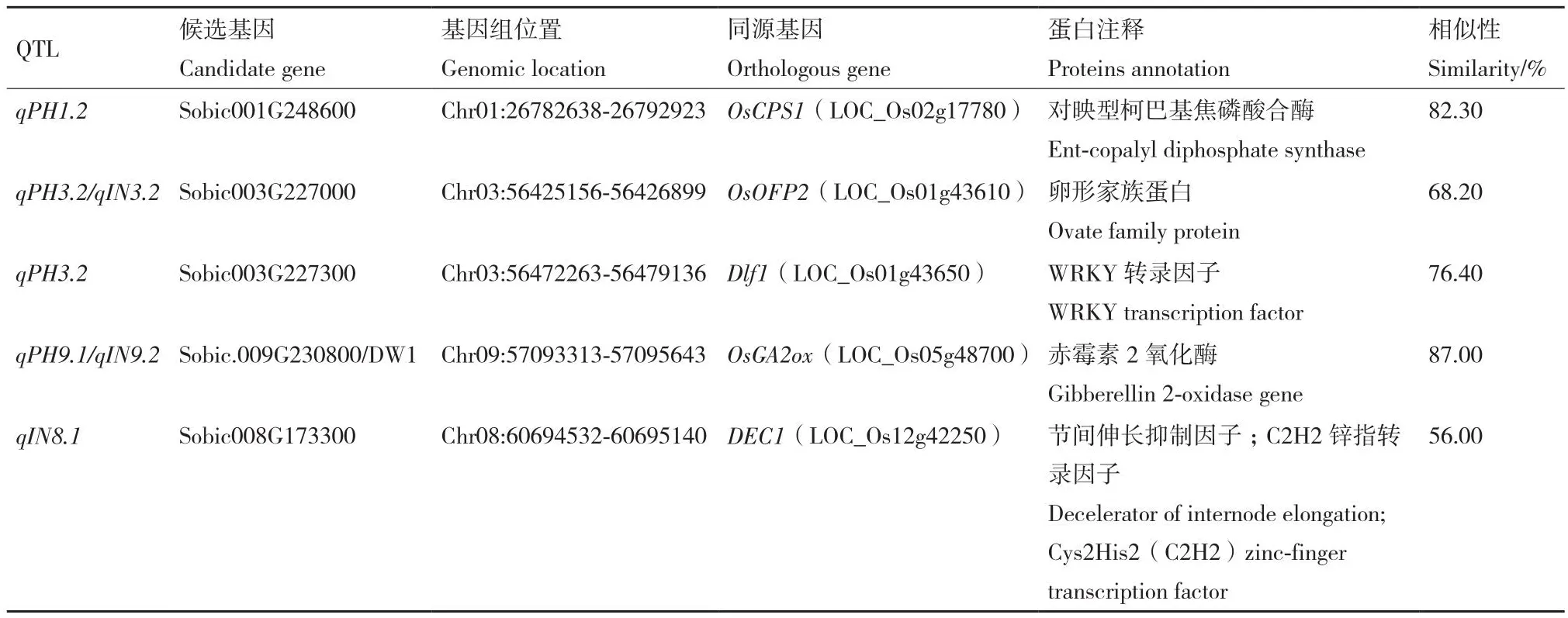
表5 重要QTL区间候选基因功能注释Table 5 Functional annotation of candidate genes in important QTL intervals
3 讨论
高粱是酿造贵州茅台酒等酱香型白酒的主要原料[23]。随着酱香型白酒产业的快速发展,贵州酒用高粱种植面积急剧上升。据统计,2022年贵州高粱生产面积达到约18.7万hm2,位居全国第一[24]。但是由于贵州酒用高粱品种普遍具有较高植株,不仅影响产量,也不适合机收[25],导致劳动力成本增加,影响农民种植积极性,从而影响酒用高粱供给。因此,开展贵州酒用高粱株高基础研究,为品种遗传改良提供依据,对白酒产业的可持续性发展意义重大。
作为多基因控制和易受环境影响的数量性状,多年多点的表型鉴定和高密度遗传图谱的构建是准确挖掘影响株高相关性状QTL的关键因素[26]。本研究对BTx623和红缨子构建的RIL群体株高和节间数于2020年在贵阳、安顺和乐东,以及2021年在贵阳和安顺5个环境条件下进行表型鉴定。相关性分析显示不同环境下相同性状之间或不同性状之间均存在极显著的正相关关系,株高与茎节数显著相关,这与Zhao等[27]和Zou等[9]研究结果一致。而利用GBS技术构建的连锁图谱,比SSR等分子标记图谱有更高的密度,具有更好的基因挖掘能力[11]。
利用Super-GBS技术构建的高密度遗传图谱,本研究定位了10个重要QTL位点在多个环境中或不同性状间都被重复检测到,其中,8个QTL位点(qPH1.2、qIN3.1、qPH4.1/qIN4.1、qIN8.1、qPH9.1/qIN9.1和qIN9.3)与已报道株高相关性状位点一致。位于第1染色体的株高QTL(qPH1.2,25.81-27.00 Mb)与前人利用2个RIL群体(M35-1/B35和BTx623/IS3620C)报道的株高QTL定位结果一致[6,28],在其置信区间的基因Sobic001G248600与水稻OsCPS1同源,编码对映型柯巴基焦磷酸合酶,参与水稻赤霉素生物合成进而影响株高[29],而且其同源物也会对玉米株高产生较大影响[30]。位于第3染色体上与茎节数相关的QTL(qIN3.1,53.08-55.06 Mb)与来源于M71/SS79的RIL的株高QTL位置重叠[31]。在3个环境中,位于第4染色体的QTL(1.48-1.53 Mb)被检测到与株高(qPH4.1)和节间数(qIN4.1)相关,与Liu等[32]利用RIL群体(Tx623/S.virgatum)定位的株高QTL位置一致。此外,在8号和9号染色体上检测到与茎节数相关的3个QTL中,qIN8.1和qIN9.1与利用SAP群体的GWAS定位结果相近[27],qIN9.3与2个RIL群体(M35-1/B35和BTx3197/Rio)定位的株高QTL结果一致[6,33]。而位于qIN8.1置信区间上的Sobic008G173300,与水稻的DEC1同源,编码C2H2锌指转录因子,在水稻和大麦植株体内过表达该基因会强烈抑制株高和节间伸长[34]。然而,位于第3染色体上的一个重要染色体区段(56.08-57.95 Mb)包含2个相邻的QTL位点(qPH3.1和qPH3.2/qIN3.2),在高粱中还未见报道与株高相关性状有关。qPH3.1(56.08-56.14 Mb)和qPH3.2(56.41-56.90 Mb)与株高相关,qIN3.2(56.23-57.32 Mb)与节间数相关。qPH3.2/qIN3.2位点所在56.23-56.90 Mb区间包含了2个候选基因Sobic003G227000和Sobic003G227300。Sobic003G227000与水稻基因OsOFP2同源,编码卵形家族,能够介导KNOXBELL功能,参与多个生长发育过程(如维管束发育)[35];而Sobic003G227300与水稻基因Dlf1同源,负责编码WRKY转录因子调节Ehd2/RID1/Osld1表达,从而参与控制水稻花期和株高[36]。上述2个QTL位点在其他高粱株高的QTL定位中未见报道的可能原因有2个:一是目前株高QTL定位群体的亲本多来源于国外甜高粱品种,而本研究的定位群体亲本之一——酒用糯高粱品种红缨子中可能携带不一样的株高基因;另一个原因是应用Sup-GBS标记技术构建高密度图谱,能够定位到更多的株高相关的QTL,而目前报道的高粱株高QTL定位多采用SSR标记[6,31-32,37]。
与水稻[38]、玉米[39]和小麦[40]等主要粮食作物相比,影响高粱株高的功能基因挖掘研究还存在较大差异[41]。截至目前,已报道控制高粱株高的主效基因有4个(Dw1-Dw4),除了位于第6染色体的Dw4以外,其余3个已被克隆[42]。本研究在3个环境中均检测到位于第9染色体上的一个重要区段(54.55-58.47 Mb)与株高(qPH9.1)和茎节数(qIN9.2)相关,正好与Dw1(57.09-57.10 Mb)的位置一致[14]。但是Dw2、Dw3和Dw4没有被检测到,可能的原因是亲本BTx623和红缨子在这3个基因上没有多态性。此外,对本研究发现的4个影响株高潜在的候选基因进一步开展克隆和功能分析,研究遗传调控机制,开发功能标记,有望为高粱株高分子育种打下基础。
4 结论
构建了包含205个RILs(Bx623/红缨子)的SNP高密度遗传连锁图谱。在5个环境下共定位到7个影响株高和11个影响节间数的QTL。在多个环境或2个性状中均检测到10个重要QTL位点,分别位于第1、3、4、8和9染色体,包含了已经克隆的Dw1位点,同时鉴定出4个与株高和节间数相关的候选基因,与控制水稻株高相关基因同源。
猜你喜欢
古今农业(2022年2期)2022-08-15
青年文学家(2022年1期)2022-03-11
今日农业(2021年20期)2021-11-26
农村百事通(2019年17期)2019-10-08
科学之谜(2019年3期)2019-03-28
科学之谜(2018年8期)2018-09-29
现代农业科技(2017年1期)2017-03-06
江苏农业科学(2016年8期)2017-02-15
江苏农业科学(2016年8期)2017-02-15
恋爱婚姻家庭·养生版(2016年9期)2016-09-07
