
QuEChERS EMR Lipid 净化结合同位素稀释-超高效液相色谱-串联质谱法同时测定畜肉中17 种β-受体激动剂
2023-08-15 01:10:34毛锐肖全伟
食品工业科技 2023年15期
毛锐,林 浩,刘 川,姚 静,肖全伟,戴 琴
(成都市食品检验研究院,国家市场监督管理总局重点实验室(营养与健康化学计量及应用),四川成都 611130)
β-受体激动剂(俗称“瘦肉精”),是一类具有促肾上腺素功能的人工合成化合物,在医学上主要用于治疗支气管哮喘、阻塞性肺炎等病症[1−2]。该类化合物具有苯乙醇胺母核,根据苯环上取代基不同,可分为苯酚型(非诺特罗、特布他林、莱克多巴胺、沙丁胺醇、福莫特罗等)和苯胺型(克伦特罗、马布特罗、溴布特罗、马喷特罗、克仑潘特等)[3]。β-受体激动剂具有营养物质“再分配效应”,可促进脂肪分解和蛋白质合成,显著提高家畜瘦肉率[4],常被不良商家非法用于家畜饲养。人食入β-受体激动剂残留的畜肉制品后可出现心慌、心悸、恶心、呕吐等症状,造成急、慢性中毒,全国食品安全整顿工作办公室发布的整顿办函〔2010〕50 号已明确将β-受体激动剂类药物列入食品中可能违法添加的非食用物质名单。
目前β-受体激动剂的常见检测方法有高效液相色谱法(HPLC)[5]、液相色谱-串联质谱法(HPLC-MS/MS)[6−10]、气相色谱-串联质谱法(GC-MS/MS)[11]、酶联免疫分析法(ELISA)[12−13]。β-受体激动剂代谢后残留于动物体内的含量非常低,HPLC 检出限难以满足检测要求;GC-MS/MS 测定时需要衍生化,操作繁琐,影响检测效率;ELISA 专属性差,检测中易出现假阳性;相比之下HPLC-MS/MS 无需衍生,灵敏度高、选择性强,已成为国内外β-受体激动剂定量分析的主要检测手段。
苯酚型β-受体激动剂在代谢过程中轭合作用较强,进入动物体内后极易与硫酸或葡萄糖醛酸形成轭合物[14−15],因此在建立苯酚型β-受体激动剂多残留分析方法时,需先对样品进行酶解,将待测物从轭合状态转化为游离态后再进行提取净化。畜肉含有丰富的蛋白质,脂肪含量高,样品基质复杂,常见的净化方式主要有固相萃取法[16−17]与QuEChERS 净化法[18−19],QuEChERS 法无淋洗、洗脱步骤,与固相萃取法相比更简便、快速,净化时仅选择性吸附杂质,不会造成目标分析物损失,在畜肉中β-受体激动剂的检测上应用越来越广泛。
增强型脂质去除填料EMR-Lipid 是一种改良的QuEChERS 技术,能高选择性地去除基质中的脂类杂质,与传统QuEChERS 法相比更适用于脂肪、蛋白质含量较高的样品,方法稳定,重现性好。目前已有文献采用QuEChERS EMR-Lipid 净化法[20]进行畜肉中β-受体激动剂的检测,但在定量时多数文献采用基质匹配标准曲线法[21]进行结果校正,实际检测工作中发现,基质曲线样品与待测样品中蛋白质、脂肪等组成不完全相同,β-受体激动剂在不同样品中的基质效应存在差异,校正效果有限,影响测定结果的准确性。同位素内标与待测物性质相近,二者在前处理及离子化过程中受到的影响基本一致,响应比值稳定[22],采用同位素内标法定量比基质匹配标准曲线法更加准确,但现有文献[23]在内标品种的选择上有较大局限性,非同位素内标与待测物在前处理及离子化过程中受到的影响存在差异,对测定结果的校正效果不佳。
为提高β-受体激动剂定量分析的准确性,本研究将QuEChERS EMR-Lipid 净化法与同位素内标稀释技术相结合,尽可能多地采用与待测物相对应的同位素内标进行校正,建立了畜肉中17 种β-受体激动剂药物残留的液相色谱-串联质谱(UPLC-MS/MS)检测方法,在简化前处理步骤、提高检测结果准确性方面具有显著优势,可满足食品安全风险监测精准定量的技术要求。
1 材料与方法
1.1 材料与仪器
牛肉、猪肉、羊肉样品 购自当地市场,用料理机搅碎备用;克仑潘特盐酸盐、羟甲基克伦特罗、喷布特罗、盐酸克仑特罗、硫酸特布他林、莱克多巴胺盐酸盐、富马酸福莫特罗、沙丁胺醇、盐酸班布特罗、非诺特罗盐酸盐标准品 纯度均≥96%,德国Dr.Ehrenstorfer 公司;马喷特罗盐酸盐、盐酸溴布特罗、苯乙醇胺A、氯丙那林、妥布特罗盐酸盐、盐酸马布特罗、西布特罗标准品 纯度均≥99%,德国WITEGA Laboratorien Berlin-Adlershof 公司;马布特罗-D9盐酸盐、班布特罗-D9盐酸盐、妥布特罗-D9盐酸盐、氯丙那林-D7、喷布特罗-D9盐酸盐、特布他林-D9醋酸盐半水化合物、溴布特罗-D9、苯乙醇胺A-D3、西布特罗-D9标准品 纯度均≥99%,德国WITEGA Laboratorien Berlin-Adlershof 公司;莱克多巴胺-D3盐酸盐标准品 纯度97.0%,加拿大C/D/N Isotopes Inc 公司;克仑特罗-D9、沙丁胺醇-D3标准品 浓度:100 μg/mL,德国Dr.Ehrenstorfer公司;甲醇、乙腈、甲酸 色谱纯,赛默飞世尔科技(中国)有限公司;乙酸铵 色谱纯,天津市科密欧化学试剂有限公司;实验用水 超纯水,由Milli-Q 型超纯水仪制备;β-葡萄糖醛苷酶/芳基硫酸酯酶>100.000 Units/mL 上海安谱实验科技股份有限公司;QuEChERS dSPE EMR-Lipid、QuEChERS Final Polish EMR-Lipid 安捷伦科技有限公司。
AB SCIEX QTRAP 5500 三重四极杆串联质谱仪 配有电喷雾离子源(ESI),美国应用生物系统公司;ACQUITYTM UPLC I-Class 超高效液相色谱仪美国沃特世科技公司;CORTECSTMUPLC®C18柱(3.0 mm×100 mm,1.6 μm)、ACQUITY UPLC BEH C18柱(3.0 mm×100 mm,1.7 μm)美国沃特世科技公司;Eppendorf Centrifuge 5810 R 型高速冷冻离心机 德国艾本德股份公司;IKA VORTEX 3 旋涡混匀器 德国艾卡公司;Heidolph 涡旋振荡器 德国海道尔夫公司;Mettler-Toledo ME204 型电子分析天平 瑞士梅特勒-托利多集团;BP211D 型电子分析天平 德国赛多利斯集团;Milli-Q 超纯水仪 美国默克集团。
1.2 实验方法
1.2.1 标准溶液的配制 混合标准溶液的配制:分别称取适量β-受体激动剂标准品,用甲醇溶解,配制成1 mg/mL 的单一标准储备液,于−20 ℃冰箱中储存。取适量各标准储备液用甲醇稀释得17 种β-受体激动剂的混合标准溶液。
混合内标溶液的配制:分别称取适量马布特罗-D9盐酸盐、班布特罗-D9盐酸盐、妥布特罗-D9盐酸盐、氯丙那林-D7、喷布特罗-D9盐酸盐、特布他林-D9醋酸盐半水化合物、溴布特罗-D9、苯乙醇胺AD3、西布特罗-D9、莱克多巴胺-D3盐酸盐标准品,用甲醇溶解,配制成0.5 mg/mL 的单一内标储备液,于-20 ℃冰箱中储存。取适量上述内标储备液及克仑特罗-D9、沙丁胺醇-D3标准品(浓度100 μg/mL),用甲醇稀释得混合内标溶液。
标准曲线工作液的配制:分别准确量取适量β-受体激动剂混合标准溶液及混合内标溶液,用10%甲醇水溶液逐级稀释,配制得浓度为0、0.5、1、2、5、10、20 ng/mL 的标准曲线工作液,内标浓度2 ng/mL,临用现配。
1.2.2 样品前处理
1.2.2.1 提取 称取均匀样品2 g(精确到0.01 g)置50 mL 离心管中,加入0.2 mol/L 乙酸铵缓冲溶液(pH5.2)5 mL、β-葡萄糖醛苷酶/芳基硫酸酯酶100 μL,2000 r/min 涡旋振荡1 min,于37 ℃避光水浴2 h。取出后放置至室温,加入100 ng/mL 混合内标溶液40 μL、5%甲酸乙腈15 mL,2000 r/min 涡旋振荡提取20 min,于4 ℃下以9500 r/min 离心5 min,待净化。
1.2.2.2 净化 在装有QuEChERS dSPE EMR-Lipid粉末的离心管中加入5 mmol/L 乙酸铵溶液5 mL,涡旋30 s 使EMR-Lipid 充分混合。将1.2.2.1 中离心后的上清液全部倒入此管,涡旋1 min,于4 ℃下以9500 r/min 离心5 min。精密吸取上清液8 mL 置另一50 mL 离心管中,倒入Final Drying Pouches-MgSO4粉末(约3.5 g),迅速振摇,涡旋1 min,于4 ℃下以9500 r/min 离心5 min。精密吸取3 mL 上清液置离心管中,在50 ℃水浴下氮气吹干,加入0.4 mL 10%甲醇水,涡旋30 s 复溶,过0.22 μm滤膜,供液相色谱-串联质谱仪测定[24]。
1.2.3 色谱条件 CORTECS™ UPLC®C18柱(3.0 mm×100 mm,1.6 μm);柱温:35 ℃;流速:0.2 mL/min;进样量5.0 μL;流动相A 为5%甲醇水溶液(含0.1%甲酸),B 为甲醇;梯度洗脱程序:0~8.00 min,10%~30% B;8.00~15.00 min,30%~95%B;15.00~18.00 min,95% B;18.00~19.00 min,95%~10% B;19.00~20.00 min,10% B。
1.2.4 质谱条件 电喷雾离子源(ESI):正离子模式;扫描方式:分时段多反应监测(Scheduled MRM,sMRM),监测窗口(MRM detection window):60 s,扫描时间(Target scan time):0.3 s。电喷雾电压:5500 V;雾化气(GS1)压力:60 psi;辅助气(GS2)压力:60 psi;气帘气(CUR)压力:20 psi;离子源温度:450 ℃;碰撞池入口电压:10 V;碰撞池出口电压:13 V。
1.3 数据处理
采用Analyst 1.6.3 软件采集数据,MultiQuant 3.0.2 软件建立标准曲线、计算结果,Microsoft Excel 2013 处理数据及绘制图表。本文基质效应和绝对萃取回收率均采用三个平行样品计算,以平均值±标准差表示。
2 结果与分析
2.1 仪器条件的优化
2.1.1 色谱条件的优化β-受体激动剂是在苯乙醇胺母核上修饰得到的系列化合物,结构相近,分离难度大。本文比较了CORTECS™ UPLC®C18柱(3.0 mm×100 mm,1.6 μm)和ACQUITY UPLC BEH C18柱(3.0 mm×100 mm,1.7 μm)对17 种β-受体激动剂的分离效果,妥布特罗、溴布特罗、马布特罗、克仑潘特因极性相近,在BEH C18柱上难以分离,换用粒径更小的实心核颗粒CORTECS™ UPLC®C18柱分离效果更好,且各化合物峰形尖锐(见图1),因此选用CORTECS™ UPLC®C18柱分离17 种β-受体激动剂。
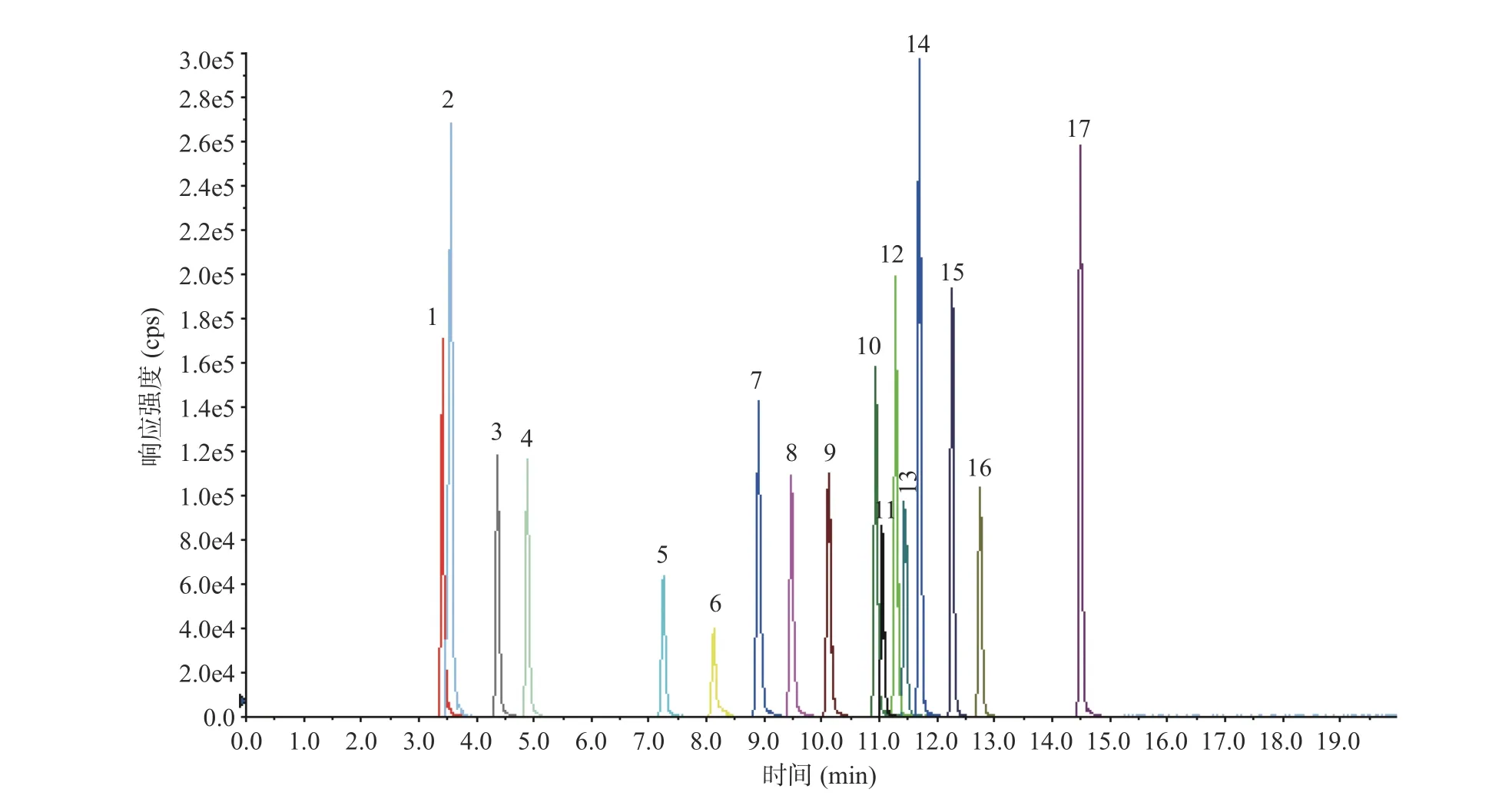
图1 17 种β-受体激动剂的提取离子流色谱图Fig.1 Extracted ion chromatogram of 17 β-receptor agonists
在相同梯度条件下比较了甲醇水、乙腈水两种流动相体系的分离效果,结果发现乙腈较甲醇有更强的洗脱能力,在乙腈水系统下β-受体激动剂出峰快,分离度更差,因而选用甲醇水系统。电喷雾离子源正离子模式下,流动相中加入适量甲酸可增强待测组分离子化效率,提高检测灵敏度,本文以甲醇-0.1%甲酸水作为流动相,调整梯度洗脱程序,尽可能使各组分基线分离,减少组分间离子化竞争。在优化后的色谱条件下(见1.2.3)17 种β-受体激动剂分离度良好,均可获得满意的检测灵敏度,优化后的谱图见图1。
2.1.2 质谱条件的优化 电喷雾离子源正离子模式下,用流动注射泵将浓度为200 ng/mL 的17 种β-受体激动剂混合标准溶液以5 μL/min 的流速注入质谱仪,通过Q1全扫描,找出准确的母离子峰,再对母离子施加一定的碰撞能量进行MS2扫描,获得各自的二级碎片离子,选取2 个信号较强且干扰小的碎片离子与其母离子组成监测离子对。在MRM 模式下,分别对去簇电压(DP)、碰撞能量(CE)等质谱参数进行优化。为了提高方法灵敏度,本研究根据17 种β-受体激动剂的保留时间,采用智能时间分段的方法进行多反应监测扫描(Scheduled MRM,sMRM),最终确定的离子对信息及质谱参数见表1。sMRM数据采集模式可根据每个化合物的保留时间自动调整监测窗口,每对离子均有最恰当的循环时间和驻留时间,能够显著提高检测方法灵敏度[25−26]。

表1 17 种β-受体激动剂的质谱参数Table 1 Mass spectrometric parameters of 17 β-receptor agonists
2.2 酶解条件的确定
GB/T 22286-2008[27]、GB/T 21313-2007[28]测定畜肉中β-受体激动剂时需先将样品酶解过夜,俞晓兰等[29]研究表明β-受体激动剂在37 ℃酶解2 h 后水解作用已基本完成,继续延长酶解时间对提取效率的影响不显著,且随水解时间延长,以特布他林为代表的部分β-受体激动剂水解产物不稳定导致测定结果降低。本文参考该文献将样品于37 ℃酶解2 h 后再用QuEChERS EMR-Lipid 净化,与β-受体激动剂日常监管常用的检测方法[27−28]相比,在保证测定结果准确性的基础上缩短了前处理时间,提高了样品检测效率。
2.3 同位素内标的选择
本文考察了牛肉、猪肉、羊肉加标样品前处理后17 种β-受体激动剂的绝对萃取回收率,结果见图2,班布特罗、克伦特罗、羟甲基克伦特罗、氯丙那林、马布特罗、妥布特罗、溴布特罗、苯乙醇胺A、马喷特罗、克仑潘特绝对萃取回收率大于60%,其余7 种绝对萃取回收率在46.3%~58.9%之间。同位素内标与待测物化学结构、理化性质相似,在前处理过程中受到的影响基本一致,二者响应比值稳定,在绝对萃取回收率不高的情况下仍能保证测定结果的准确性。为实现精准定量分析,本研究在经费许可的情况下,尽可能多地购置与待测物相对应的同位素内标,定量分析时均以自身同位素内标进行计算。非诺特罗、福莫特罗、克仑潘特、马喷特罗、羟甲基克伦特罗未购得对应的同位素内标,非诺特罗、福莫特罗与莱克多巴胺同属苯酚型β-受体激动剂,三者具有相近的绝对萃取回收率与基质效应(见图2、图3),故以莱克多巴胺-D3作为非诺特罗、福莫特罗的内标。克仑潘特、马喷特罗、羟甲基克伦特罗均为苯胺型化合物,绝对萃取回收率与同为苯胺型的马布特罗、克伦特罗相近(见图2),但克伦特罗和羟甲基克伦特罗基质抑制效应较强(见图3),以克伦特罗-D9作羟甲基克伦特罗的内标可补偿基质抑制效应对羟甲基克伦特罗的影响,故选择克伦特罗-D9为羟甲基克伦特罗的内标,马布特罗-D9为克仑潘特、马喷特罗的内标。
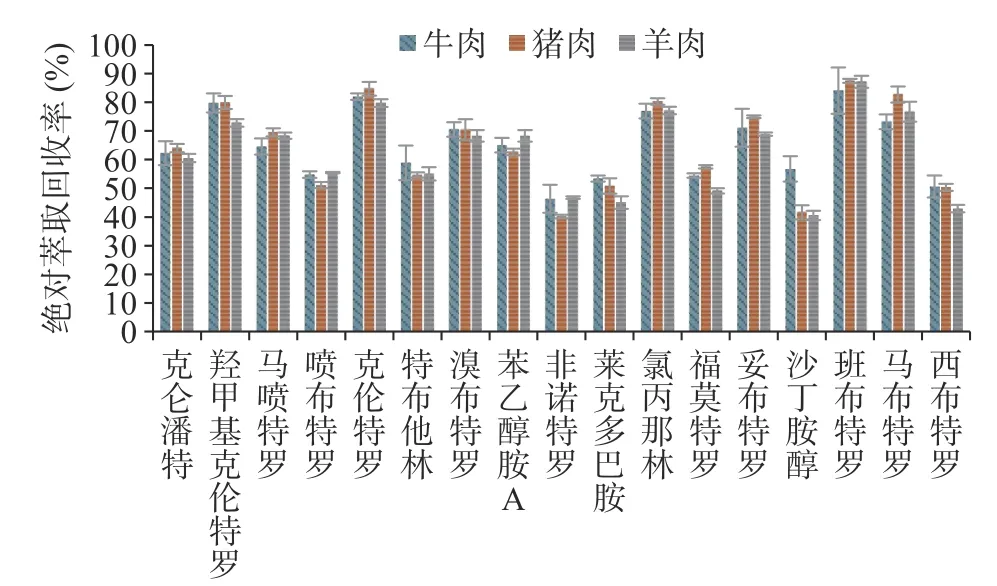
图2 牛肉、猪肉、羊肉样品中17 种β-受体激动剂的绝对萃取回收率Fig.2 Absolute extraction recovery of 17 β-receptor agonists in beef,pork and mutton

图3 牛肉、猪肉、羊肉样品中17 种β-受体激动剂的基质效应Fig.3 Matrix effect of 17 β-receptor agonists in beef,pork and mutton
2.4 基质效应的考察
基质效应(ME)计算方法见公式(1),式中A1为待测物溶于有机溶剂中的峰面积,A2为待测物溶于空白基质溶液中的峰面积,ME 在±20%以内为可接受范围[30−31]。
基质效应结果见图3,牛肉、猪肉、羊肉对17 种β-受体激动剂均呈现出基质抑制效应,其中克伦特罗、特布他林ME 绝对值大于20%,具有明显的基质抑制效应,其余15 种β-受体激动剂ME 绝对值在5.16%~17.9%之间,基质效应较弱,表明QuEChERS EMR-Lipid 法净化效果良好。克伦特罗、特布他林定量分析时均以自身同位素标志物作内标,补偿了基质抑制效应对测定结果的影响,提高了定量准确性。
2.5 QuEChERS 法与MCX 固相萃取法净化及提取效果对比
以牛肉为例,比较QuEChERS 法与MCX 法对17种β-受体激动剂的净化效果。基质效应对比结果见图4,两种前处理方法均呈现基质抑制效应,大多数β-受体激动剂基质效应不明显,仅有QuEChERS法克伦特罗和特布他林的ME 绝对值略大于20%,表明两种前处理方法均有较强的基质干扰去除效果。

图4 牛肉样品QuEChERS 法与MCX 法基质效应对比图Fig.4 Matrix effect comparison between QuEChERS method and MCX method in beef samples
牛肉样品QuEChERS 法与MCX 法的绝对萃取回收率对比结果见图5,17 种β-受体激动剂在QuEChERS 法中的萃取回收率普遍高于MCX 法。MCX 柱是混合型阳离子交换柱,具有反相和阳离子交换双重保留性能,对非极性的碱性化合物具有更高的选择性。MCX 柱净化需经历上样、淋洗、洗脱过程,17 种β-受体激动剂母体结构中苯环上取代基不同,极性与碱性各有差异,在MCX 柱上呈现出不同的保留特性,同一净化条件下理化性质差异大的化合物易在淋洗中损失或在洗脱中流出不完全,难以兼顾所有化合物的绝对萃取回收率,MCX 净化下喷布特罗、苯乙醇胺A、福莫特罗的绝对萃取回收率仅有12.3%、16.4%、27.2%。QuEChERS 法使用的EMRLipid 是一种新型吸附材料,该吸附剂结合体积排阻和疏水作用两种机制,能高选择性地去除基质中的脂类杂质,净化时仅有杂质吸附与脱水过程,不会造成目标分析物损失,对17 种β-受体激动剂能提供更为均衡的绝对萃取回收率,最低绝对萃取回收率也能达到46.3%,与MCX 法相比更适用于多残留的同时检测,因而本研究选用QuEChERS EMR-Lipid 法进行样品前处理。
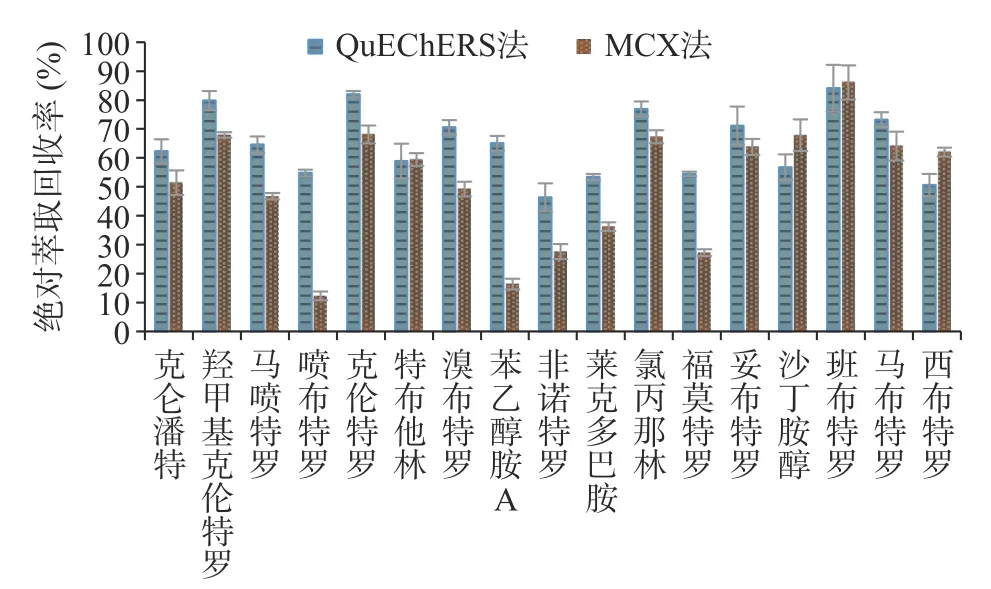
图5 牛肉样品QuEChERS 法与MCX 法绝对萃取回收率对比图Fig.5 Absolute extraction recovery comparison between QuEChERS method and MCX method in beef samples
2.6 方法学考察
2.6.1 线性范围与检出限 取标准曲线系列工作液进样测定,以标准品与相应内标的峰面积比为纵坐标,以标准品浓度为横坐标,绘制标准曲线,得到线性回归方程。采用逐步稀释法,以3 倍信噪比(S/N)为检出限,10 倍信噪比(S/N)为定量限,结果见表2。17种β-受体激动剂在0~20 ng/mL 范围内线性关系良好,相关系数均大于0.99,检出限为0.01~0.09 μg/kg,定量限为0.05~0.31 μg/kg,可满足检测要求。

表2 17 种β-受体激动剂线性范围、回归方程、相关系数、检出限及定量限Table 2 Linear equation,correlation coefficient,LOD,LOQ of 17 β-receptor agonists
2.6.2 回收率与精密度 选择猪肉、牛肉、羊肉阴性样品,以低(0.5 μg/kg)、中(2.0 μg/kg)、高(5.0 μg/kg)三个浓度进行加标回收试验,每个浓度水平各6 份,按照1.2.2 项下方法处理样品后进样测定,计算方法回收率与精密度,结果如表3 所示,3 个浓度水平下,猪肉样品中17 种β-受体激动剂的平均回收率为83.1%~109.3%,相对标准偏差为0.8%~9.8%,牛肉样品中17 种β-受体激动剂的平均回收率为81.7%~111.8%,相对标准偏差为1.0%~10.2%,羊肉样品中17 种β受体激动剂的平均回收率为86.5%~111.6%,相对标准偏差为1.0%~10.3%。本方法具有较好的准确度和精密度,可满足畜肉中17 种β-受体激动剂的检测要求。12 种以自身同位素标志物作内标的β-受体激动剂平均回收率在91.4%~111.8%之间,与部分已有方法[10,23]相比准确度更高,定量更为精准可靠。

表3 17 种β-受体激动剂回收率及精密度(n=6)Table 3 Recoveries and RSD of 17 β-receptor agonists (n=6)
2.7 实际样品测定
采用本文建立的方法对100 批市售生鲜畜肉中17 种β-受体激动剂进行检测,其中猪肉31 批,牛肉38 批,羊肉31 批,结果均为未检出,暂未发现生鲜畜肉中β-受体激动剂残留风险,与GB/T 22286-2008《动物源性食品中多种β-受体激动剂残留量的测定 液相色谱串联质谱法》检测结果一致。
3 结论
本研究采用QuEChERS EMR-Lipid 净化法及同位素稀释-超高效液相色谱-串联质谱技术建立了畜肉中17 种β-受体激动剂药物残留的检测方法。酶解2 h 后的样品经QuEChERS EMR-Lipid 净化,前处理步骤简便,净化效果良好,有效消除了基质效应影响,同时缩短酶解时间,提高了样品检测效率。采用同位素内标法定量,补偿了样品前处理过程对测定结果的影响,17 种β-受体激动剂在0~20 ng/mL 范围内线性关系良好,3 个加标浓度水平下平均回收率为81.7%~111.8%,RSD 为0.8%~10.3%,检出限为0.01~0.09 μg/kg,定量限为0.05~0.31 μg/kg,定量精准可靠,适用于大批量畜肉样品中多种β-受体激动剂药物残留的同时检测,为动物源性食品风险监测提供了技术支持,具有广阔的应用前景。
猜你喜欢
农药科学与管理(2023年9期)2023-12-14 11:44:14
好孩子画报(2020年2期)2020-03-30 03:47:05
知音海外版(上半月)(2018年5期)2018-05-23 03:11:20
创新作文(小学版)(2017年22期)2017-04-04 02:17:16
合成化学(2015年9期)2016-01-17 08:57:02
应用化工(2014年1期)2014-08-16 13:34:08
食品工业科技(2014年7期)2014-03-11 18:14:48
郑州大学学报(理学版)(2014年2期)2014-03-01 04:20:57
精细石油化工(2014年2期)2014-02-28 02:04:35
食品科学(2013年14期)2013-03-11 18:25:08
