
急性髓系白血病中组蛋白乙酰化研究进展
2023-08-12 02:07:02范世洁王曼曼黄赞
生物化工 2023年3期
范世洁,王曼曼,黄赞*
(1.武汉大学 生命科学学院,湖北武汉 430072;2.安徽中医药大学 新安医学教育部重点实验室,安徽合肥 230031)
急性髓系白血病(Acute Myeloid Leukemia,AML)是一类起源于造血干/祖细胞恶性扩增的血液肿瘤,表现为克隆性增殖的异常分化或低分化造血细胞在骨髓、血液和其他组织中的广泛浸润。随着人们对AML 认识的不断深入,AML 治疗在近20 年来有了长足的进步,总体治愈率在30%~40%,部分类型白血病如急性早幼粒白血病的5 年生存率甚至可以达到90%[1-2]。然而,AML 在细胞遗传学上有显著异质性,并且存在多种不良预后类型的AML,其临床治疗效果依然不容乐观。
驱动AML 发生的遗传变异涵盖了点突变、缺失、扩增、基因易位等。肿瘤基因组图谱研究显示这些遗传变异在功能上主要分为9 类[3],其中DNA 甲基化和表观遗传修饰因子是AML 重现突变驱动基因中的一大类,是影响白血病起始、发生和发展的关键因素之一。多种DNA 甲基化/去甲基化酶以及识别蛋白在造血和白血病发生中的生理病理功能已有广泛研究,相应抑制剂也在白血病临床治疗中得到应用[4]。组蛋白修饰是表观遗传的另一个重要内容,主要包括甲基化、乙酰化、磷酸化等,参与调控染色质结构和基因表达,在多种生物学过程中发挥重要作用[5]。相对其他表观遗传修饰,组蛋白乙酰化在白血病发生、发展和治疗中的作用及意义并未得到充分认识,靶向组蛋白乙酰化治疗AML 的策略依然面临诸多挑战。
1 组蛋白乙酰化
组蛋白乙酰化修饰发生在组蛋白N 端尾部的多个赖氨酸残基上,影响染色质构象,导致转录复合物的募集改变,并最终调节基因表达。作为DNA 包装的支柱,组蛋白在基因表达调控中起着核心作用,并有助于正常细胞功能的发挥;异常组蛋白表观遗传共价修饰可能是控制细胞中功能失调相关基因表达的因素,尤其在癌细胞中。组蛋白H3 的N 端赖氨酸乙酰化修饰与转录激活密切相关,是一个高度调节和可逆的过程,由特定的蛋白质家族介导。组蛋白乙酰化的动态平衡及细胞生物学功能主要依赖由乙酰辅酶A 辅助的乙酰转移酶(HAT)作为组蛋乙酰化“编写器”来乙酰化组蛋白,由NAD+或锌辅助因子辅助的脱乙酰基酶(HDAC)作为“橡皮擦”来移除组蛋白的乙酰化修饰,以及HAT 及相关蛋白(如GCN5L2)、组蛋白甲基转移酶(例如ASH1L 和MLL)、溴域蛋白(BET)蛋白家族、转录共激活因子、核支架蛋白PBRM1 等作为表观遗传“阅读器”来识别赖氨酸上的乙酰基[6]。
本系统的开发软件为Delphi 7.0;数据库管理系统为oracle 9i;辅助处理系统为office2003Photoshop 6.0。计算机操作系统选用:Windows9x2000NTXP或更高级的操作系统都可以安全运行本系统。
2 组蛋白乙酰化调控失衡参与AML 发生
异常的组蛋白乙酰化修饰被证明与AML 密切相关,且AML 病人往往呈现低水平组蛋白乙酰化[7-8]。组蛋白乙酰化“编写器”“阅读器”等介导的乙酰化调控失衡是AML 发生、发展中不可忽视的因素。
2.1 组蛋白乙酰化“编写器”参与AML 发生
HAT 家族的多个成员的遗传变异导致HAT 活性失调是白血病发生的驱动突变,其中融合HAT 基因的染色体重排是导致AML 组蛋白乙酰化调控异常最直接的因素。这些AML 亚类携带t(11;16)(q23;p13.3)、t(8;16)(p11;p13)和t(8;22)(p11;q13)的 嵌合转录本,分别编码MLL-CREBBP、MOZ-CREBBP和MOZ-EP300 融合蛋白,引起AML 的驱动遗传变异[9]。HAT 与髓系恶变的相关性并不局限在融合伴侣,MLL-AF9 和NUP98-HOXA9 驱动的白血病增殖依赖MOF 乙酰转移酶活性,无乙酰辅酶A 结合能力的MOF 突变体会显著影响融合蛋白转化骨髓细胞的克隆形成效率[10];NUP98-HOXA9 和CBFB-MYH11招募乙酰化转移酶引起特异位点组蛋白乙酰化修饰水平升高是白血病发生的重要机制[11-12]。混合谱系白血病(Mixed Lineage Leukemia,MLL)中DOT1L 靶基因的H3K79me2 修饰能够促进组蛋白乙酰化修饰,并在MLL 白血病中发挥重要作用[13]。组蛋白乙酰化转移酶HBO1 在维持白血病干细胞中发挥关键作用,HBO1 缺失能延长MLL-AF9 小鼠白血病模型的存活时间[14]。NUP98-HBO1 融合蛋白则是造成慢性粒细胞白血病的因素[15]。HAT 在AML 中的相关性还超越乙酰转移酶活性。例如,香豆素(Celastrol)通过破坏Myb 类转录因子家族和EP300 融合蛋白之间蛋白质相互作用抑制Myb 激活的靶基因,这种抑制作用与EP300 乙酰转移酶活性抑制不相关;同时抑制EP300酶活性及其与Myb 的相互作用也可以协同诱导体外模型中的白血病细胞髓系分化[16]。这些研究表明HAT 同时具备酶活和支架功能,显示异常组蛋白乙酰化在白血病发生发展中的关键作用。
长叶山兰发现于贵州雷公山,生境海拔1 988 m,生于路边林下潮湿沟谷,伴生种有竹根七、楼梯草、水芹、黔川乌头等。2015年9月20日引种保存于贵阳药用植物园,2018年5月首次开花后进行了鉴定,凭证标本:HXQ2015092034HT。
按:“荐祀”,犹祭祀。“荐祀”,其他文献用例亦富,例如《风俗通义》卷第五:“还历乡里,荐祀祖考。”《樊川文集》卷第六《三子言性辩》:“梁武帝起为梁国者,以笋脯麦牲为荐祀之礼。”《震川先生集》卷三十《告昆山县城隍神文》:“奕奕新庙,荐祀馨香。”《牧斋初学集》卷第五十三《明故陕西革昌府通判钱君墓志铭》:“汉有良吏,乐府流传。弦歌荐祀,安阳亭西。”《乐府诗集》卷第八《豫章府君登歌》:“嘉乐在庭,荐祀在堂。”“荐祀”一词,《汉语大词典》未收。
Lys-CoA 和H3-CoA-20 是最早的双底物类似物,选择性抑制p300 和PCAF。无心果酸(anacardic acid)、藤黄酚(garcinol)和姜黄素(curcumin)是有效的天然p300 和PCAF 抑制剂。其他化合物如藤黄酚类似物、γ-丁内酯MB-3、异噻唑酮(isothiazolones)和喹啉(quinoline)衍生物可抑制特定的HAT 成员并有效阻断某些实体肿瘤细胞系的增殖,但其作用机理仍未阐明[55]。噻唑的衍生物CPTH6 能抑制GCN5 和PCAF 活性,降低AML 细胞整体的乙酰化水平,诱导细胞凋亡和分化[56]。MOZ 和MORF 双价抑制剂能与乙酰辅酶A 强力竞争,在小鼠模型中抑制白血病细胞增殖[57]。HAT 选择性抑制剂C646(吡唑啉酮-呋喃化合物)特异靶向CREBBP 的催化活性及相关乙酰转移酶EP300(也称为KAT3B),能显著降低组蛋白乙酰化水平,在10 个测试的AML 组织培养细胞系中显著降低了8 个细胞系的增殖,并在多个亚型的AML 中展现出良好的临床前治疗效果[58]。乙酰赖氨酸竞争性抑制剂特异性抑制CBP/p300 的BRD 结构域,显著降低AML 细胞增殖,破坏MLL-AF9 驱动的白血病起始细胞的自我更新和疾病进展[59]。双氟尼醛(diflunisal)与乙酰辅酶A 竞争CBP/p300 活性催化位点,有效阻止组蛋白乙酰化,抑制依赖p300 的AML-ETO 白血病细胞增殖[60]。然而,HAT 抑制剂的选择性和生物利用度低,使得这些抑制剂难以进入临床实验。
针对于我国会计师事务所的实际发展情况,事务所业务多层次发展必将成为一种大趋势,所以,现阶段的事务所应当分析多方面要素的影响,适时更新经营理念、与时俱进,发挥国家政策、服务质量等因素对事务所的贡献,提高自身核心竞争力。与此同时,把握住主观因素对事务所自身的影响,探寻出适合自身情况的多层次发展对策,进而使会计师事务所实现更健康、更良性的发展、同时亦为我国注册会计师行业的可持续发展做出长足贡献。
2.2 AML 与识别组蛋白乙酰化赖氨酸的表观遗传“阅读器”相关
干扰组蛋白乙酰化“阅读器”的功能是另外一种针对组蛋白乙酰化的干预方式。多种BET 蛋白抑制剂能模拟乙酰化赖氨酸阻止BET 识别组蛋白乙酰化赖氨酸,其功能已经在包括白血病在内的几种肿瘤类型中进行了测试,见表2。
其中,小分子JQ1(硫代三唑并二嗪)和I-BET151(3,5-二甲基异恶唑衍生物)在体外和体内均可有效诱导MLL 重排白血病细胞的细胞周期阻滞和凋亡[65,80]。这些抑制剂的作用是将BRD4 从调节元件中置换出来,并在特定癌基因(包括c-MYC、BCL2 和CDK6)的位点上阻断RNA Pol II 介导的转录延伸。同样,BET 抑制剂OTX015(JQ1 衍生物)显示出在多种白血病细胞类型中诱导凋亡的能力[81]。目前多项BET 抑制剂已经开展临床试验(表3)。
寒假的第二天,天气清朗。冬日的阳光白白的,好像力度不够,晒在人身上,也还觉得暖洋洋的。陈浩的家和我家在同一条街上,吃完早饭,我就出发去他家了。
3 靶向组蛋白乙酰化治疗AML 的策略
基于对组蛋白乙酰化在白血病发生中的功能认识,干预组蛋白乙酰化是潜在白血病治疗策略。通常干扰组蛋白乙酰化的策略有3 种:破坏组蛋白乙酰化“阅读器”BET 与HAT 蛋白复合物缔合的复合物抑制,抑制HAT 乙酰基转移酶活性或HDAC 去乙酰化酶活性的抑制剂,以及阻止BET 蛋白与乙酰化的组蛋白结合的变构位点抑制。
3.1 靶向组蛋白乙酰化的HAT 抑制剂
到目前为止,经调查,仅有少数组蛋白乙酰转移酶抑制剂(HAT 抑制剂)被鉴定出来(表1),其在白血病治疗中尚未得到广泛研究。
DPF2(Double Plant Homeodomain Finger 2)是高度进化保守的d4 蛋白家族成员,含有植物同源结构域(PHD 手指结构域),能结合组蛋白的乙酰化赖氨酸组。有研究显示DPF2 能抑制造血干/祖细胞髓系分化,抑制AML 细胞增殖,而组蛋白结合缺陷的DPF2 突变体则丧失抑制髓系分化的能力[25-26]。这些研究表明DPF2 可能在白血病发生中发挥重要作用。
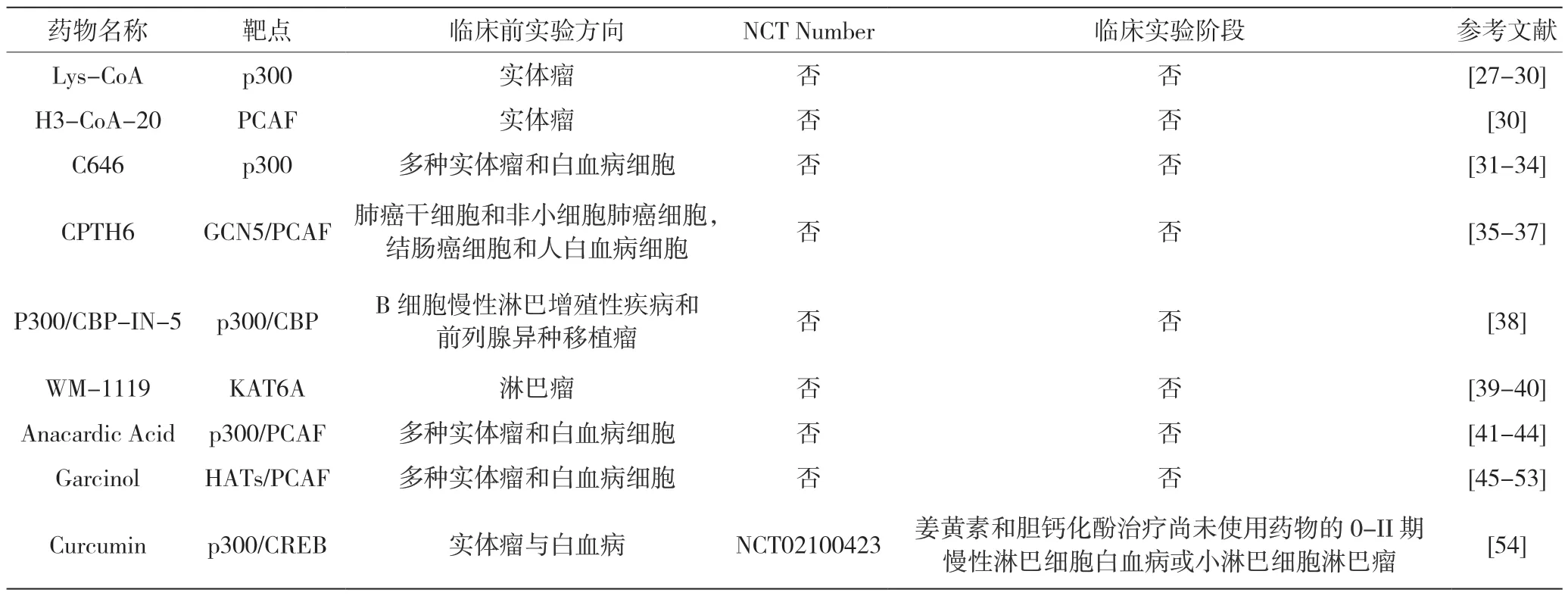
表1 HAT 抑制剂的临床前研究进展
一项细胞系蛋白质谱研究表明H3K9 甲基化,H3K14、H3K18 和H3K23 乙酰化水平以及潜在的H4K20 甲基化水平升高与白血病细胞阿霉素耐药性有关,涉及转录激活和沉默[17]。同时,非APL 类型的AML 细胞对ATRA 等诱导分化剂诱导分化治疗具有抗性,这种抗性与组蛋白乙酰化转移酶GCN5 和PCAF 相关:GCN5 通过调控H3K9 乙酰化控制干性和白血病相关基因表达,阻止ATRA 诱导白血病细胞髓系分化[18]。同时,PCAF 能诱导ATRA 靶基因启动子区域的H3 组蛋白乙酰化,是全反式维甲酸诱导AML 细胞向粒细胞终末分化所必需的[19]。
3.2 靶向组蛋白乙酰化的HDAC 抑制剂
相较于HAT 抑制剂,组蛋白去乙酰化酶抑制剂(HDAC 抑制剂)则显示出良好的治疗功效。目前,美国FDA 已经批准了多种HDAC 抑制剂用于治疗多种白血病,如伏立诺他(Vorinostat)、帕比司他(Panobinostat)和贝利司他(Belinostat)等。多项研究评估了HDAC 抑制剂在AML 中的疗效,并取得了可喜的结果。在t(8;21)驱动的AML 的体内模型中,给药Panobinostat 触发病原性AML1/ETO 融合蛋白的蛋白酶体降解,从而导致了终末髓样分化,并在小鼠中获得了出色的存活率[61]。然而,Panobinostat 的活性仅限于AML/ETO 的这一AML 亚类,无法推广到更多AML 类型中。这些结果表明无论是HAT 还是HDAC 的抑制剂都有抑制AML 的效果,提示AML 细胞中的HAT 和HDAC 活性平衡可能是决定AML 发生和恶变的重要因素,总的组蛋白乙酰化水平则影响较小;采用何种方法干预AML 细胞中组蛋白乙酰化稳态以及AML 对相应干预方法的敏感性可能取决于AML 起源的特定遗传变异背景。
3.3 靶向表观遗传的BET 抑制剂
这些表观遗传“阅读器”具有专门的结构域识别核小体乙酰化修饰,充当其他调控因子的募集支架。BET 蛋白是一类乙酰结合蛋白,它们通过在赖氨酸残基处结合组蛋白和非组蛋白的乙酰基团发挥作用。其中,BRD4 是发育和细胞分化中转录网络的关键调节因子,也是驱动肿瘤细胞异常转录程序的关键参与者;BRD4 结合并识别称为“超级增强子”的H3K27乙酰化的特殊区域,控制多个谱系特异性基因表达,并可能被肿瘤细胞劫持以表达关键的癌基因[20]。在AML 细胞中,BRD4 维持c-MYC 的表达以促进白血病细胞自我更新[21]。除了组蛋白乙酰化染色质“阅读器”活性以外,研究还显示BRD4 具有HAT 活性,可导致染色质松弛并在物种间保持保守[22]。

表2 BET 蛋白抑制剂的临床前研究
含 有YEATS 结 构 域 的ENL(Eleven-Nineteen Leukemia)和AF9 是另一类组蛋白乙酰化“阅读器”[23]。YEATS 域倾向识别部分乙酰化组蛋白肽,包括H3K27ac、H3K9 和H3K18 乙酰化。t(11;19)(q23;p13.1)染色体易位导致ENL 与MLL(KMT2A)基因融合,(9;11)(q22;q23)染色体易位导致AF9 与MLL 基因融合,随之表达的MLL-ENL 和MLL-AF9嵌合蛋白是白血病的驱动遗传变异,导致基因表达失调,促进急性白血病的发生。ENL 耗竭具有抗白血病效应,包括增加终末髓样分化和抑制体外和体内白血病细胞增殖。ENL 与乙酰化组蛋白H3 结合,并与H3K27 和H3K9 乙酰化共同定位在对白血病至关重要的活跃转录基因的启动子上[24]。
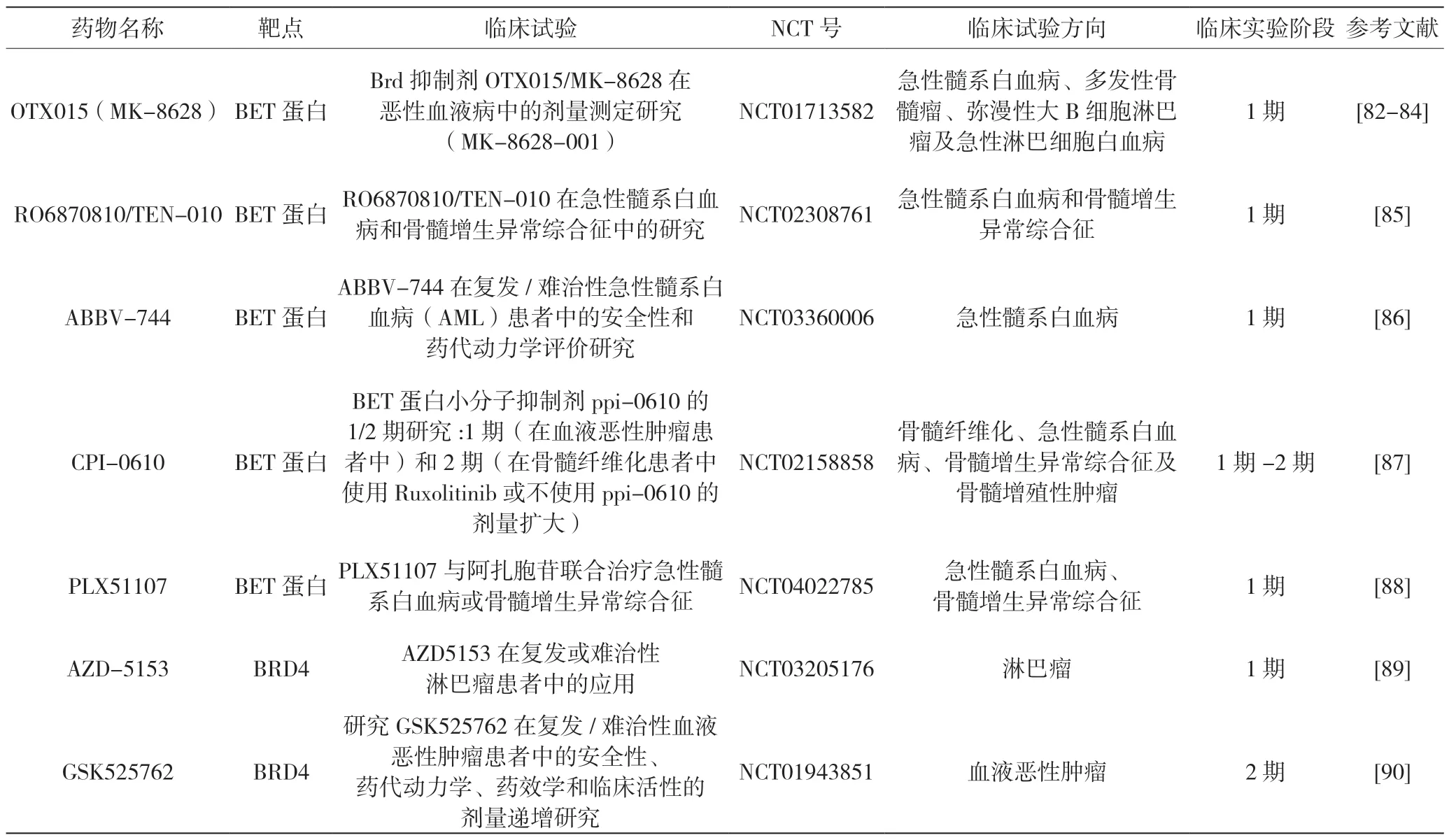
表3 BET 蛋白抑制剂的临床研究进展
小分子RO6870810/TEN-010(JQ1 类似物)已在难治性AML 和骨髓增生异常综合征(Myelodysplastic Syndromes,MDS)进行了Ⅰ期试验(NCT02308761),BET 抑制剂CPI-0610(氯苯甲基乙二唑苯扎西汀)的Ⅱ期试验测试了其与芦可替尼(ruxolitinib)联用治疗骨髓纤维化(NCT02158858)的效果。在同一管线上,BRD4 抑制剂GSK525762 进入了难治性血液恶性肿瘤复发患者的早期临床试验。BET 抑制剂BI-894999(属于三唑吡嗪类)与其他BET 抑制剂在结构上截然不同,虽然调控与JQ1 相同的基因,但在杀死原代AML 细胞和异种移植模型的AML 细胞方面更出色,与CDK9(转录延伸复合物的组成部分)抑制剂的组合可大大增强其抗肿瘤作用[91]。虽然靶向BET 蛋白在白血病治疗方面取得相当好的进展,但仍然缺乏有价值的BET 转基因动物模型来阐明BET 抑制剂的毒性作用及其发挥作用的机理。
4 ANP32A 调控AML 组蛋白乙酰化
ANP32A 属于ANP32(Acid Nuclear Phosphoprotein,32 kDa)基因家族成员,是组蛋白乙酰转移酶抑制复合物的组分,能与非修饰的组蛋白N 末端结合,体外抑制组蛋白H3 乙酰化修饰。除了结合组蛋白抑制乙酰化以外,早期研究显示ANP32A 还具有多种功能,包括抑制去磷酸化酶PP2A、激活凋亡小体、mRNA转运、结合微管相关蛋白(Microtubule-Associated Protein,MAP)等[92]。最近的研究表明ANP32A 调控ATM(Ataxia Telangiectasia-Mutated)表达阻断软骨、脑和骨的氧化应激,而ANP32A 的种属差异是决定甲型流感病毒聚合酶宿主局限性的原因[93-94]。
研究表明,ANP32A 是白血病发生的重要调控因子,是影响AML 细胞组蛋白乙酰化的重要因素。ANP32A 耗竭能显著抑制AML 病人原代细胞和AML细胞系的增殖与克隆形成能力,ANP32A 缺失会损害MLL-AF9 体外转化小鼠骨髓细胞的能力[95-96]。深入的机制研究显示,尽管ANP32A 不具备HAT 活性,但AML细胞中高表达的ANP32A促进H3组蛋白乙酰化,进而调控脂代谢基因表达,推动白血病发生。这个研究结果与最初体外ANP32A 抑制组蛋白乙酰化功能截然相反,提示ANP32A 在体内对组蛋白乙酰化修饰的作用可能与其招募的相互作用蛋白有关。为了进一步阐明ANP32A 调控组蛋白乙酰化的机制,研究人员进一步干预ANP32A 和H3 组蛋白的相互作用,结果显示破坏ANP32A 和H3 组蛋白的相互作用能抑制AML 细胞增殖[97]。表明ANP32A 是AML 组蛋白乙酰化的重要辅助因子,通过ANP32A 干预组蛋白乙酰化有望成为AML 治疗的新策略。
别名地丁、地丁草、紫花地丁、小鸡菜、扁豆秧,为罂粟科植物紫堇的干燥全草,夏季花果期采收,除去杂质,晒干。主要分布于辽宁、河北、内蒙古、山东、山西、陕西、甘肃、宁夏等。
5 展望
尽管组蛋白乙酰化在基因表达激活以及开放染色质结构中的功能已经非常明确,但其在白血病发生中的功能及其在白血病治疗中的潜在作用并未得到充分阐释,特别是白血病发生过程中组蛋白乙酰化稳态的动态变化规律有待进一步阐明,而通过研究不同类型AML 病人原代细胞和构建相应的动物模型可以进一步揭示其中的变化规律。现有的研究表明抑制组蛋白乙酰化和抑制组蛋白去乙酰化都能达到白血病治疗效果,提示白血病细胞中组蛋白乙酰化的稳态失衡是关键因素,而组蛋白乙酰化水平本身的影响较小。因此,后续研究有必要阐明组蛋白乙酰化水平与AML 各个亚类的相关性,明确不同亚类AML 对组蛋白乙酰化抑制剂或组蛋白去乙酰化抑制剂敏感性的差异,从而科学判断抑制剂的选择。
另外,发展靶向组蛋白乙酰化的AML 治疗依然面临多重挑战。尽管其机制仍未得到充分探索,但已有证据表明白血病对表观遗传疗法也会产生抗性,其中最典型的是对BET 抑制剂的抗性。因此,开展多种疗法组合是提高AML 治疗效果的有效途径,可以选择与常规化疗、靶向疗法、其他表观遗传疗法甚至免疫疗法进行组合优化,以期达到提高疗效的目的[98]。有报道指出许多HAT 小分子抑制剂生物利用度低和特异性差,且具有抗氧化或不稳定性等细胞内特性,极大阻碍了HAT 抑制剂在临床上的应用。因此,持续研究和测试优化影响HAT 和组蛋白乙酰化“阅读器”的化合物,将有助于使用此类分子的疗法设计以用于临床试验。但是,靶向组蛋白乙酰化的AML 治疗的更大挑战在于靶向组蛋白乙酰化系统的复杂性,因为该系统是大规模整合的多个蛋白质和多个单一底物的复合物,而靶向复合物中单个蛋白的单个结构域小分子很可能无效,尤其是在复合物中多个蛋白存在功能冗余的情况下。不可忽视的是,该系统中存在类似ANP32A 的“辅助”蛋白作为功能刺激蛋白影响HAT 活性及其生理病理功能的发挥。所以,未来开展系统性的研究鉴定这类相关功能蛋白,可能为该干预策略拓展思路,并提供更多的靶向分子,以期提高治疗效果。
猜你喜欢
军事文摘(2024年2期)2024-01-10 01:59:00
分子催化(2022年1期)2022-11-02 07:11:08
中国生物化学与分子生物学报(2022年8期)2022-09-08 00:39:50
河南畜牧兽医(2017年12期)2017-11-13 04:05:18
中国组织化学与细胞化学杂志(2016年4期)2016-02-27 11:15:53
中国科技信息(2015年6期)2015-11-10 03:35:44
云南中医学院学报(2015年2期)2015-07-31 18:11:59
化学反应工程与工艺(2015年1期)2015-04-16 03:06:16
中国当代医药(2015年16期)2015-03-01 02:03:13
中国塑料(2014年4期)2014-10-17 03:00:50
