
变应性鼻炎中lncRNA-miRNA-mRNA网络构建与相关程序性细胞死亡基因的分析
2023-07-08 04:32王宇婷姜辉王嘉玺
中国耳鼻咽喉颅底外科杂志 2023年3期
王宇婷,姜辉,王嘉玺
(北京中医药大学东方医院 耳鼻咽喉科,北京 100078)
变应性鼻炎(allergic rhinitis, AR) 是耳鼻咽喉科常见疾病之一,属于机体免疫系统疾病。然而其发病机制的研究仍有不明之处,导致其诊断及针对性治疗存在一定的困难。因此,我们迫切的需要进一步研究,并且对AR的生物学机制有更深入的了解。非编码RNA(ncRNA),包括microRNA(miRNA)、长链非编码RNA(lncRNA)和环状RNA(circRNA),在表观遗传学、转录调控和转录后调控中发挥重要作用[1]。越来越多的研究表明lncRNA广泛参与AR的发生发展过程。lncRNA-miRNA-mRNA所形成的内源竞争RNA(competing endogenous RNA,ceRNA)调控网络在免疫疾病中已有广泛研究。然而,ceRNA网络与AR的发生发展之间的关系仍不清楚。
Zhang等[2]研究表明FOXD3-AS1在AR患者鼻黏膜中下调,其抑制鼻黏膜上皮细胞(nasal mucosa epithelial cells,NEC)中白细胞介素-25(interleukin-25,IL-25)的表达和分泌,从而调节AR中的辅助性T淋巴细胞2(helper T lymphocytes,Th2)的功能。然而,也有证据表明lncRNA不独立发挥调节功能,而通过与某些ncRNA竞争性结合相互作用,构建动态的调节网络,这以前被称为竞争。ceRNA网络被认为是解释转录后调控的重要机制[3]。有学者[4]研究发现在AR患者的鼻黏膜组织和IL-13处理的NEC中LINC00632表达下调,并负调节miR-498,从而降低其靶基因白介素1受体拮抗剂(interleukin1 recepter antagonist,IL1RN)的表达,以此来调节巨噬细胞集落刺激因子、嗜酸性粒细胞趋化因子等的表达。然而AR的研究不仅限于此,考虑到程序性细胞死亡(programmed cell death,PCD)在AR中的重要作用,我们根据构建的ceRNA网络进一步对AR的调控机制进行探讨。
本研究选取AR患者与正常人全血样本进行RNA测序,构建lncRNA介导的ceRNA网络,对网络中的mRNA进行基因本体(gene ontology,GO)和KEGG功能富集分析。然后,我们将ceRNA网络中差异表达的mRNA与PCD相关的基因进行交互作用,并构建lncRNA介导的ceRNA网络中的PCD相关调控网络,分析相关基因的靶器官定位。该研究进一步深入挖掘AR发病的新的分子机制,特别是lncRNA介导的ceRNA调控网络与程序性死亡之间的相互作用。
1 材料和方法
lncRNA介导的ceRNA网络(lncRNA-miRNA-mRNA)的构建及相关PCD基因分析流程如图1所示。

图1 ceRNA网络的构建与分析流程图 注:AR(变应性鼻炎);GO(基因本体)。下同。
1.1 患者和样本收集
1.1.1 一般资料 本研究纳入4例AR患者,其中男1例,女3例;平均年龄29.3岁,平均病程2.4年。对照组4例,男2例,女2例;平均年龄30.3岁。两组均于2021年3月—2021年4月就诊于北京中医药大学东方医院耳鼻咽喉科门诊,8例患者均久居于北京市。所有对照组均为变应原特异性IgE阴性。研究对象均进行常规检查及静脉血采集,所有的血液样本都取自肘部静脉。该研究所用标本通过北京中医药大学东方医院伦理委员会批准,并征得患者知情同意。
1.1.2 纳入及排除标准 纳入标准: ①年龄18~55岁;②符合国内《变应性鼻炎诊断和治疗指南》(2015年)中制定的AR诊断标准[5];③免疫印迹法进行的血清变应原特异性IgE(specific IgE, sIgE)检测阳性,主要对尘螨、动物皮屑、蟑螂或霉菌过敏;④AR病史≥1年;⑤同意参与本研究并签署知情同意书者。同时符合以上5条者方可纳入。
排除标准:①近2周内患呼吸道感染或急性鼻窦炎;②慢性鼻窦炎病史或影像学显示鼻窦炎;鼻腔手术史或鼻腔器质性病变;③近3个月曾接受过西药抗过敏治疗、中药治疗,可能影响本临床研究的观察治疗测定者;④合并全身免疫系统疾病、严重呼吸系统疾病、血液循环系统疾病、造血系统疾病者;⑤恶性肿瘤患者;⑥精神病患者;⑦妊娠期妇女;符合上述任意一条者均不能入组。
1.1.3 全血样本采集和保存 应用PAXgene血液RNA管(preanagen,Venlo,荷兰)采集全血2.5 mL,PAXgene采血管内含能够稳定体内基因转录性状的抗凝剂以避免RNA快速降解,稳定细胞内RNA。将PAXgene试管小心倒置8~10次,可确保管内保护剂和血液充分混合均匀,在室温下放置2 h后,大体积干冰运输至深圳微科盟科技集团有限公司实验室进行检测,以减少RNA降解。
1.2 RNA测序分析
1.2.1 全血总RNA提取 在RNA提取前,从干冰中取出试管后,室温解冻并在孵育2 h。RNA纯化按照PAXgene血液RNA试剂盒手册中的方案进行。利用NanoDrop2000仪(NanoDrop技术公司,威尔明顿,DE,美国)对所提 RNA 的浓度和纯度进行检测,琼脂糖凝胶电泳测RNA完整性,电泳图片显示3条清晰的条带(28 s/18 s/5 s),RNA完整性良好。Qubit测定浓度。单次建库要求RNA总量≥1 ug,浓度≥100 ng/μL,OD260/280≥1.8,OD260/230≥1.0。
1.2.2 RNA测序实验 测序实验采用Illumina Truseq TM RNA sample prep Kit方法进行链特异文库构建,包括去除rRNA,RNA片段化,反转成cDNA,连接adaptor即在双链的 cDNA 在3’末端加上一个 A 碱基,连接Y字形的接头,UNG酶消化cDNA二链,文库富集即PCR扩增,纯化得到最终文库,并由illumina平台上机测序。lncRNA是一类转录本长度超过200 nt,不编码 lncRNA的RNA分子。我们基于转录组拼接结果,根据 lncRNA 的结构特点以及不编码 lncRNA 的功能特点,设置一系列严格的筛选条件,将筛选所得lncRNA进行后续分析。使用StringTie-eB软件对lncRNA的转录本进行定量分析,得到各样本的转录本readcount 及FPKM信息。同时对测序结果中的mRNA及miRNA进行定量分析,得到readcount 及FPKM值。
1.3 筛选差异表达lncRNA、miRNA和mRNA
采用R软件中的“DESeq2”软件包,对AR患者与对照组之间的lncRNA、miRNA和mRNA进行差异表达分析。用P<0.05和|log2FC|>1作为基因表达显著性差异的筛选阈值。通过使用R软件中的“ggplot2”、“pheatmap”和“ggrepel”软件包生成热图和火山图来进行差异基因的可视化。
1.4 lncRNA-miRNA-mRNA网络的构建
lncRNA-miRNA-mRNA网络基于ceRNA假设构建:①应用miRmap, miRanda, miRDB, TargetScan和MitarBase数据库提取miRNA-mRNA的相互作用信息,应用 StarBase数据库提取 miRNA-lncRNA相互作用的信息;②如果lncRNA和mRNA都被同一个miRNA靶向,并且该miRNA与lncRNA及mRNA表达负相关,则该lncRNA-miRNA-mRNA组被确定为共表达竞争三联体,故构建相应的ceRNA调控网络。应用cytoscape 3.7.1软件对ceRNA调控网络进行可视化。
1.5 GO和KEGG功能富集分析
对上述步骤中得到的ceRNA网络中的mRNA进行潜在的生物学功能富集分析。GO富集分析分为分子功能(molecular function, MF)、生物过程(biological process,BP)和细胞组成(cellular component,CC)3个部分;KEGG分析是应用KEGG(京都基因和基因组百科全书)数据库进行mRNA相关通路的富集分析,利用R软件中的“clusterprofiler”包预测ceRNA网络中的mRNA潜在的生物学功能。P<0.05为差异具有统计学意义。KEGG分析应用R软件中“GOplot”包筛选GO中3个部分的前20个基因集绘制柱状图进行可视化;并筛选具有显著差异的KEGG基因集进行圈图绘制。
1.6 ceRNA网络中PCD相关基因的筛选
首先genecards数据库(https://www.genecards.org)中与PCD相关的基因,包括“程序性细胞死亡”、“细胞凋亡”、“细胞焦亡”相关基因。然后,将ceRNA网络中差异表达的mRNA与PCD、细胞凋亡和细胞焦亡相关的基因相互取交集。结果通过在线工具Venny 2.1.0(http://bioinfogp.cnb.csic.es/tools/venny/index.html)生成的Venny图进行可视化。
1.7 PCD相关基因的ceRNA网络构建
依据ceRNA假设构建PCD相关lncRNA-miRNA-mRNA网络,并应用Cytoscape 3.7.1软件对ceRNA调控网络进行可视化。
1.8 ceRNA网络中mRNA与靶器官网络的构建
AR作为免疫系统疾病其发病机制尚有不明之处,可能有多个组织和器官参与疾病的发生进展。因此,应用BioGPS数据库(https://biogps.org)在器官组织水平上检测每个mRNA的表达谱信息[6]。该数据库提供了通过微阵列分析直接测量基因表达而获得的基因表达数据[7]。我们首先得到84个器官和组织中每个mRNA表达的分布数据,然后计算每个组织中每个mRNA的平均值和所有组织中的总体平均值[8]。最后,提取mRNA表达量高于总体平均值的相关器官和组织中的前14个靶器官与mRNA通过Cytoscape 3.7.1建立mRNA-靶器官定位网络。
2 结果
2.1 差异表达的lncRNA、miRNA及mRNA的鉴定
对AR组及对照组全血样本测序,并对已知序列的lncRNA、miRNA和mRNA进行定量分析,以P<0.05和|log2FC|>1为阈值筛选差异表达的lncRNA、miRNA和mRNA。共鉴定出200个差异表达lncRNA(98个上调,102个下调);201个差异表达mRNA(92个上调,109个下调);14个差异表达miRNA(7个上调,7个下调)。差异表达的lncRNA、miRNA和mRNA的热图和火山图如图2所示。

图2 差异表达的lncRNA、mRNA和miRNA的火山图和聚类热图 A:差异表达lncRNA火山图; B:差异表达mRNA火山图; C:差异表达miRNA火山图; D:200个差异表达lncRNA聚类热图; E:201个差异表达mRNA聚类热图; F:14个差异表达miRNA聚类热图 注:红色表示上调的基因,蓝色表示下调的基因;浅绿色表示对照组,淡红色代表AR组。
2.2 lncRNA、miRNA、mRNA间ceRNA网络的分析
我们根据碱基序列和表达水平分别预测了lncRNA-miRNA和miRNA-mRNA对。基于ceRNA网络构建思路,在lncRNA介导的上调ceRNA网络中鉴定出4对miRNA-lncRNA和22对miRNA-mRNA,包括3个lncRNA节点、2个miRNA节点和22个mRNA节点;在lncRNA介导的下调ceRNA网络中鉴定出3对miRNA-lncRNA和24对miRNA-mRNA,2个lncRNA节点、2个miRNA节点和24个mRNA节点。见图3。

图3 lncRNA介导的ceRNA调控网络的构建 A:lncRNA介导的上调ceRNA网络; B:lncRNA介导的下调ceRNA网络 注:绿色表示LncRNA上调,红色表示LncRNA下调;淡黄色表示miRNA下调,紫色代表miRNA上调;蓝色代表mRNA上调,橘黄色代表mRNA下调。
2.3 ceRNA网络中差异表达mRNA功能富集分析
为了进一步探索lncRNA介导的ceRNA网络相关的潜在功能,应用R中的“clusterprofiler”对ceRNA网络中的mRNA进行功能富集分析。GO分析结果将显著富集(P<0.05)的前20个GO条目绘制成柱状图(图4A)。结果表明,参与ceRNA网络中的差异表达mRNA尤其富集于GO:0051924(钙离子转运的调节);GO:0070372(ERK1和ERK2级联的调节);GO:0070371(ERK1和ERK2级联);GO:0014065(磷脂酰肌醇 3-激酶信号传导)等生物学进程中。GO:0016323(基底外侧质膜);GO:0016324(顶端质膜);GO:0031253(细胞投射膜)等细胞成分中。GO:0016651[作用于 NAD(P)H的氧化还原酶活性];GO:0140375(免疫受体活性);GO:0005178(整合素结合)等分子功能。见图4B。另外KEGG通路分析表明,ceRNA网络中mRNA的功能主要富集于hsa04510(粘附斑激酶通路);hsa05235(在癌症中PD-L1的表达和PD-1检查点通路);hsa04020(钙信号通路);hsa04611(血小板活化通路);hsa04151(PI3K-Akt 信号通路);hsa04022(cGMP-PKG信号通路);hsa04621(NOD样受体信号通路);hsa04060(细胞因子与受体间相互作用);hsa04080(神经活性配体-受体相互作用)。见图4C。

图4 ceRNA网络中差异表达mRNA的功能富集分析 A:ceRNA网络中差异表达mRNA的GO分析 注:BP(生物过程);CC(细胞成分);MF(分子功能)。X轴:各GO条目;Y轴:校正过的P值(P<0.05表示显著富集)。红色表示在AR中该GO生物学功能更有可能上调,蓝色表示更有可能下调。B:筛选可能具有临床意义且显著富集的10个GO条目绘制圈图 注:蓝色点代表下调基因,红色点代表上调基因;内圈条状的高度代表富集基因的数目;内圈条状的颜色:红色表示在AR中该GO生物学功能更有可能上调,蓝色表示更有可能下调。C:筛选可能具有临床意义且显著富集的9个KEGG通路绘制圈图,用于展示基因的差异倍数与富集到的KEGG通路间的隶属关系 注:左侧代表基因,其中红色上调基因,蓝色代表下调基因;右侧代表9个KEGG通路。
2.4 ceRNA网络中PCD相关基因的鉴定及PCD相关基因的ceRNA网络分析
我们将ceRNA网络中的46个差异表达的mRNA与GeneCards数据库中检索到14 004个自噬相关基因相互作用,鉴定出ceRNA网络中存在25个自噬相关基因,于13 473个PCD相关基因相互作用,共获得25个交集基因;与184个焦亡相关基因相互作用,鉴定出1个交集基因(图5)。然后,我们对构建了AC008394.1及人类组织相容性白细胞抗原复合物P5(human histocompatibility leukocyte antigen comple P5,HCP5) 2个lncRNA所介导的ceRNA调控网络,探索其对PCD的调控作用,结果如图6所示。

图5 韦恩图,ceRNA网络中差异表达的mRNA与PCD、细胞凋亡、细胞焦亡相关的mRNA之间的相互作用 注:PCD(程序性细胞死亡)。下同。

图6 PCD相关基因ceRNA调控网络的构建 A:lncRNA介导的下调ceRNA网络; B:lncRNA介导的上调ceRNA网络 注:黄色表示lncRNA下调,红色表示lncRNA上调;浅蓝色表示miRNA上调,橘黄色代表miRNA下调;紫色代表mRNA下调,绿色代表mRNA上调。
2.5 ceRNA网络中mRNA与靶器官定位网络分析
基因表达数据是基于B和GPS数据库中的基因表达芯片分析所得。在查找相关mRNA在BioGPS数据库的表达谱中,NANOGP8和CD247没有mRNA表达谱。故我们分析了44个mRNA靶蛋白在84个组织和器官中的表达谱,选取前14个靶器官与相应的mRNA构建mRNA-靶器官定位网路,14个靶器官包括小脑、颞叶、松果体、下丘脑、背根神经节、骨髓、CD34+等以神经免疫系统为主的相关组织器官。网络中包含32个节点和328条边。大多数靶标同时在多个器官和组织中高表达,表明这些器官与PCD相关ceRNA网络密切相关,提示其相关作用机制可能与神经、免疫等功能相关。见图7。
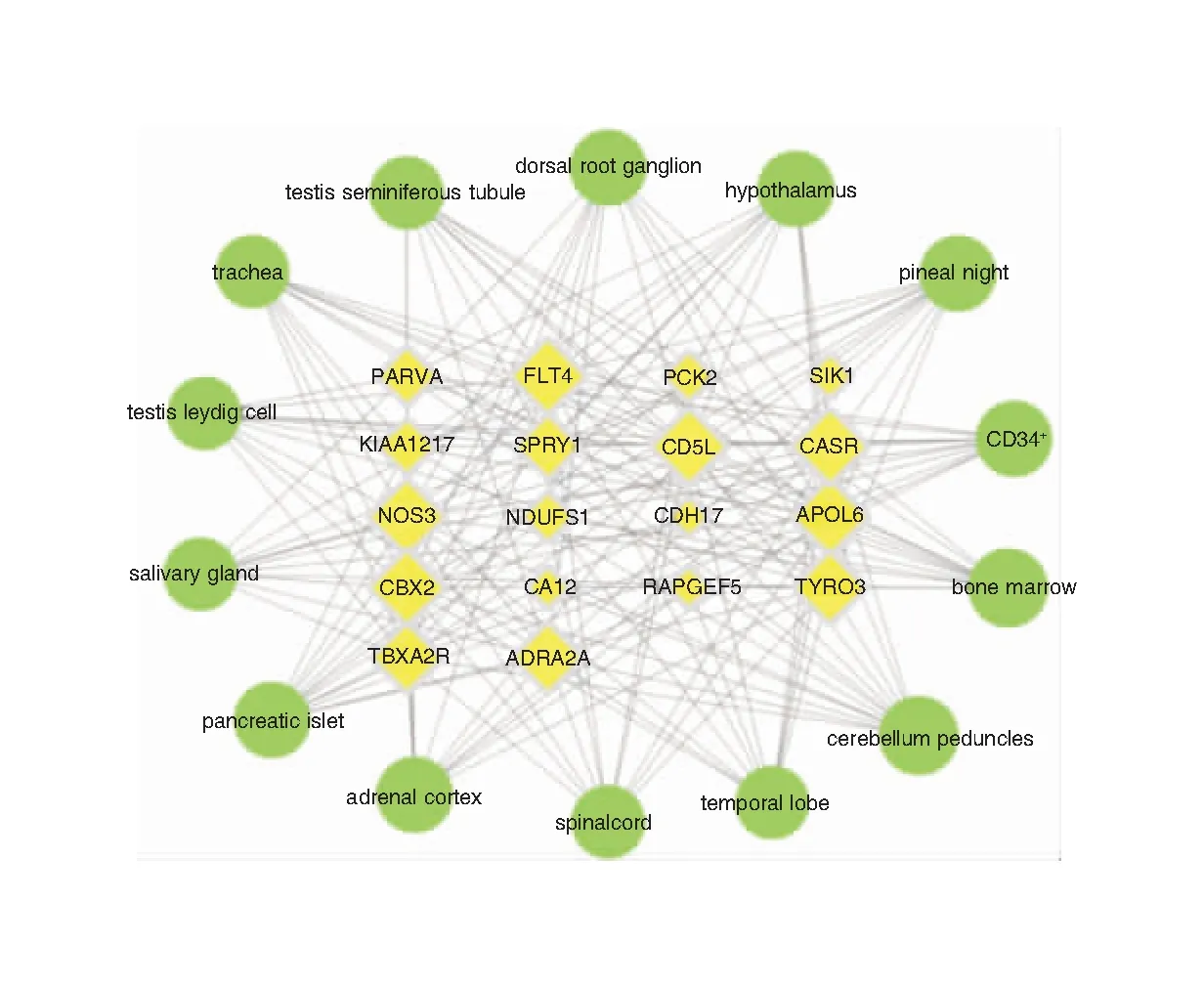
图7 ceRNA网络中mRNA与靶器官定位网络的构建 注:黄色代表基因;绿色代表靶器官。节点大小由degree值决定。
3 讨论
AR是一种复杂的疾病,随着空气质量日益恶化,人们生活工作压力的增加及饮食不规律等原因,使得患病人数逐年增加,且部分患者伴发哮喘等下呼吸道疾病,严重影响生活质量[9]。虽然常年性 AR患者较少存在生存问题,但疾病所产生的附加影响,如对情绪、睡眠及经济成本的影响仍值得我们去研究。然而, AR的分子机制尚不清楚,这为疾病的早期诊断及针对性治疗带来困难。
为了更好地认识 AR的发病机制和发现潜在的生物标志物,本研究利用转录组测序技术对其进行了定量和全面的分析,包括编码和非编码转录组。结果显示,常年性 AR患者与正常人中miRNA、lncRNA和mRNA的表达模式存在显著差异,而DEmiRNA、DElncRNA和DEmRNA的动态变化也存在显著差异。近年来,ncRNA 被发现与广泛的生物调节功能相关[10]。然而,本研究获得的DElncRNA大多功能未知,这主要是由于对它们的研究较少。因此,我们构建了常年性 AR由lncRNA介导的ceRNA调控网络。基于ceRNA理论构建了2个lncRNA-miRNA-mRNA调控网络,包括5个lncRNA、4个miRNA和46个mRNA。lncRNA AC004832.1、AC008394.1、TDRG1、HCP5和CHL1-AS1均首次在 AR患者外周血中被鉴定为差异表达的lncRNA。GO和KEGG富集分析表明,参与ceRNA网络的DEmRNA在“钙离子转运的调节”、“PI3K-Akt通路”和“免疫受体活性”等中显著富集。在ceRNA网络与PCD相关基因相互作用后,鉴定到20个lncRNA介导的ceRNA调控通路。另外,将鉴定到的ceRNA网络中的mRNA进行靶器官定位分析发现,大多数靶标在神经免疫系统为主的相关组织器官中表达。
在上述ceRNA调控通路中,有几个枢纽节点已被证明在AR及相关免疫细胞中发挥关键作用。人类主要组织相容性复合体(human major histocompatibility complex,MHC),也被称为人类白细胞抗原(human leukocyte antigen,HLA),覆盖人类基因组的0.13%,跨越6号染色体短臂6p21位置,该区域包含超过250个注释基因和假基因[11],lnRNAHCP5基因位于6p21.33区域中,人白细胞抗原HLA I类β区域内的ERV16元件中[12]。在一项关于HCP5与转录因子相互作用的研究中,Warner等[13]通过敲除NF-κB1基因失活的研究中发现,HCP5、NOD2和IL-8与细胞活力下降密切相关。同时NF-κB1(位于chr4上)调控他一些MHC基因的表达,从而影响多种生物学功能,包括与炎症疾病相关的错误激活、不适当的免疫细胞发育和细胞生长延迟。且它可以独立于大多数基因被激活或抑制。另外在探讨细胞因子对HCP5表达的影响时发现,在外周血单核细胞中IL-10能够下调HCP5的表达[14],而IL-10在AR中低表达[15],故推测HCP5可能在AR中表达增高,这与本研究结果一致。
目前对于HCP5通过ceRNA机制发挥作用的研究主要集中在肿瘤研究中,Cheng等[16]在结直肠癌的研究中,应用荧光素酶报告基因分析,结果表明miR-139-5p是HCP5的直接靶点,HCP5通过激活ZEB1并与miR-139-5p相互作用,参与上皮-间充质转化。Zhang等[17]研究发现HCP5通过靶向miR-140-5p/IGF1R通路促进透明细胞肾细胞癌的增殖和转移,HCP5作为一种相互竞争的内源性RNA,通过在ccRCC中海绵化miR-140-5p来调节IGF1R的表达。如在肺腺癌的研究中发现蛋白编码基因CTSS、FGL2和PDL2,miR-106b-5p和miR-17-5b及lncRNAHCP5形成了调控网络,从而HCP5通过与免疫检查点基因PDL2和抑制癌变的治疗靶点FGL2竞争,参与了肺癌的发生过程[18]。HCP5通过海绵化miR-22-3p、miR-186-5p和miR-216a-5p发挥ceRNA功能,激活ST6GAL2,调节滤泡性甲状腺癌细胞的增殖、迁移、侵袭性和血管生成能力,从而促进滤泡性甲状腺癌进展[19]。
ceRNA网络中的miRNA是重要的节点,Zhou等[20]通过qRT-PCR检测患者和小鼠鼻黏膜中miR-31的表达水平发现在AR患者中miR-31水平升高。Çemeci 等[21]对鼻息肉组织与邻近的正常鼻黏膜相比,发现鼻息肉组织中miR-31的平均水平升高,且在气道上皮细胞中高表达[22]。另有miR-31对免疫细胞作用机制的研究,用miR-31转染CD4+T细胞可降低IL-2和IL-4的表达水平[23],上述在 AR及气道炎症中对于miR-31的研究结果,与本研究结果相同。在花粉季节期间和之外获取SAR患者CD4+T细胞,使用微阵列分析发现miR-139为差异表达的miRNA,结合共表达分析结果显示miR-139可能通过调节FOS和JUN影响SAR[24]。另外,来自季节性AR患者的CD4+T细胞,应用芯片分析,结果提示miR-139-3p和miR-223可以发挥互补作用,靶向改变Th2细胞功能,影响Th2细胞因子的释放[25]。既往对于miR-140的研究发现,在AR患者鼻黏膜中应用qRT-PCR检测lnc-GAS5、其靶基因miR-21和miR-140及干扰素-γ、IL-2、IL-4 和 IL-10,结果显示在AR患者中lnc-GAS5升高,而 miR-21和miR-140显著下调,干扰素-γ和IL-2与lnc-GAS5呈正相,结果提示miR-140与AR的疾病风险、症状严重程度和Th1/Th2失衡相关[26]。该研究结果与本研究不同,可能为选取检测组织样本不同,使得miR-140的表达存在差异,更需要进一步的验证。
ceRNA网络中的mRNA是最终发挥效应的主要节点,既往研究发现,血栓素 A2 受体 (thromboxane A2 receptor,TBXA2R)是与慢性气道炎症有关的基因。多项研究都提示TBXA2R与呼吸道疾病哮喘间存在密切关系[27-28],且TBXA2R中的rs8113232 SNP位点与哮喘儿童鼻炎之间存在关联[29]。NOS3缺失导致卵清蛋白致敏小鼠出现中等强度的气道高反应。除了气道反应性的差异,在气道炎症程度、卵清蛋白特异性IgE水平方面没有差异[30]。
为了进一步探讨AR中差异表达的mRNA生物学功能,我们对ceRNA中46个差异表达mRNA进行了GO和KEGG通路分析,我们发现这些mRNA在“钙信号通路”和“PI3K/AKT信号通路”中显著富集,这与之前的观点一致。免疫细胞中的钙信号通路参与细胞分化,基因转录和效应器功能的调节。细胞内钙离子水平(Ca2+)的增加是由于免疫感受器的参与,例如T细胞受体,B细胞受体和Fc受体,以及趋化因子和共刺激受体。诱导淋巴细胞中细胞内Ca2+水平增加的主要途径是通过储存操作的钙进入和钙释放激活的钙通道[31]。肥大细胞通过释放组胺和炎症因子在AR的过敏性炎症中起关键作用,这一过程称为脱颗粒[32],蛋白激酶C激活和胞质Ca2+浓度升高的水平这两种途径可能共同激活肥大脱颗粒作用,故胞质Ca2+浓度增加对肥大细胞脱颗粒有重要意义[33]。Nian等[34]在研究过敏原激发的NEC与AR中肥大细胞脱颗粒之间的关联时发现,IL-33及其受体ST2的表达在NEC中升高;同时在肥大细胞中IL-33降低了LC3-Ⅰ/Ⅱ和Beclin-1水平,增加了p62水平,表明LAD2细胞的自噬受到抑制,并验证了IL-33是通过ST2/PI3K/mTOR介导的自噬来促进AR中肥大细胞的脱颗粒。此外,构建的ceRNA网络中的mRNA在“ERK1和ERK2级联”中显著富集。ERK2/ERK1(也被称为p42/p44MAPK,正式命名为MAPK1和3),是两种细胞外信号调节激酶(ERK)的异构体,属于丝裂原活化蛋白激酶家族成员[35]。Qin等[36]研究发现AR患者血清中IL-36和Th17细胞因子(IL-17和IL-23)的表达显著上调。而IL-36α可通过介导ERK和PI3K/AKT通路,显著增加Th17细胞的转录因子RORγt的表达,并上调PBMCs对IL-17和IL-23的产生。且IL-17和IL-23可以促进IL-36α的产生,形成一个正反馈回路,从而对AR发挥调控作用。另外有研究称Ras/Raf/ERK通路可诱导细胞凋亡,其作用机制包括调节Bcl-2蛋白家族控制细胞色素c的释放及促进caspase-8信号传导和激活等[37]。因此,我们有理由假设,新构建的ceRNA网络可能通过调节Ca2+转运在免疫细胞及细胞因子相互作用在AR中发挥作用。
基于对ceRNA网络中mRNA生物学功能的分析,结果提示相关机制与自噬及凋亡存在一定联系,故我们将46个mRNA与PCD相关基因相互取交集,探究特征性PCD在调节AR中发挥的作用。细胞死亡根据可控性分为PCD和非PCD两大类。炎症的发生多于PCD相关,但近年来的研究显示多种细胞死亡也受到程序性控制,多种PCD也与炎症发生相关。PCD包括凋亡、自噬、坏死性凋亡、焦亡、铁死亡及细胞衰老等形式[38]。在我们的研究中发现ceRNA网络中的mRNA主要与凋亡关系密切。凋亡在形态上会出现细胞皱缩、染色质凝集、凋亡小体形成和细胞骨架解离等特征。线粒体凋亡途径受控于BCL2蛋白家族,死亡受体途径受死亡受体的介导。而线粒体凋亡在特定条件下对炎症[39]和天然免疫[40]产生影响。既往研究发现,凋亡与AR相关,包括对中性粒细胞凋亡[41]、嗜酸性粒细胞[42]、嗜碱性粒细胞[43]和鼻上皮细胞凋亡[44]等与AR发生间关系的研究。HCP5与miR-3619-5p相互作用以调节胃癌(gastric cancer,GC)细胞的凋亡[45]。新构建的ceRNA网络中的mRNA,如巨噬细胞凋亡抑制剂又称 CD5L,主要由巨噬细胞分泌,其可在炎症反应和免疫抑制相关病理过程中重要发挥作用[46],故我们推测HCP5可能通过ceRNA机制调节细胞凋亡从而对AR产生影响。最后,我们将新构建的ceRNA中的mRNA进行靶器官定位网路分析,显示mRNA主要在神经免疫系统为主的相关组织器官中表达,提示与神经、免疫等功能有关。
本研究通过RNA测序确定了来自AR组及对照组中差异表达的lncRNA、miRNA和mRNA数据,并构建了ceRNA网络,初步探索出HCP5介导的ceRNA网络可能通过影响细胞凋亡对AR发挥作用。研究仍存在一些局限性,首先选取的样本量较少,可能会影响结果的质量,且暂未对测序结果进行相关表达量及功能验证,进一步研究应扩大样本量并进行相关试验验证。其次,研究选取外周血成分相对复杂,结合既往研究,未来可针对相关免疫细胞进行筛选和验证。这些实验可能有助于了解AR的病理机制,帮助寻找AR治疗的潜在药物靶点。
猜你喜欢
流行色(2021年8期)2021-11-09
学苑创造·A版(2020年12期)2020-01-07
中国外汇(2019年15期)2019-10-14
科学中国人(2017年36期)2017-06-09
发明与创新(2016年26期)2016-08-22
作文教学研究(2016年1期)2016-07-05
中国病理生理杂志(2015年8期)2015-12-21
西南医科大学学报(2015年1期)2015-08-22
医学研究杂志(2015年8期)2015-06-22
医学研究杂志(2015年3期)2015-06-10
