
Potocki-Lupski 综合征及重组人生长激素治疗随诊1 例报告
2023-06-30 09:26:14刘祖娴姜丽红刘戈力陈鑫王美林
天津医科大学学报 2023年3期
刘祖娴,姜丽红,刘戈力,陈鑫,王美林
(天津医科大学总医院儿科,天津 300052)
Potocki-Lupski 综合征(Potocki-Lupskisyndrome,PTLS)是由于17p11.2 区域微重复所导致的一类常染色体显性遗传疾病,重复区域大小从0.41 Mb 到19.7 Mb 不等,3.7 Mb 的微重复约占PTLS 的2/3。其主要临床表现是发育迟缓、婴儿期肌张力低下、吞咽困难导致的喂养困难、睡眠呼吸暂停、结构性心血管异常、智力障碍、行为异常、身材矮小、生长激素缺乏相关性低血糖及轻微的面部畸形等。其患病率约为1/25 000[1-2]。本文报道了1 例因身材矮小合并伴智能发育落后前来就诊的PTLS 患儿的临床特点,及其接受重组人生长激素(recombinant human growth hormone,rhGH)治疗并随访的临床资料,以提高临床医师对此病的认识及诊治水平。
1 临床资料
患儿,男,6 岁1 月龄,主因身材矮小3 年余伴智力落后于同龄儿就诊于我院儿科门诊。患儿为第1 胎,第1 产,足月自然分娩,否认窒息史。出生身长50 cm,体重3.4 kg。2 岁7 月龄时身高84 cm(-2.41 SD)。生长发育里程碑稍落后(3 个月抬头,5 个月翻身,7 个月会坐,13 个月走路,1 岁半会说简单词语,3 岁会说完整句子),智能发育落后于同龄儿。父亲身高165 cm,母亲身高163 cm,父母外貌及智能均正常。父母非近亲婚配,家族中无类似病史。查体:身高106 cm(-2.58 SD),体重17 kg(-1.79 SD),精神好,特殊面容(前额突出、眼距宽、塌鼻梁、高腭弓),肤色黑,双眼斜视、远视,鸡胸明显,脊柱、四肢无畸形,心音有力,律齐,心率88 次/min,双肺呼吸音清,未闻及干湿性啰音,腹软,肝脾肋下未触及,无压痛及反跳痛。外阴Tanner 分期Ⅰ期,阴茎3 cm,双侧睾丸均2 mL。肌力肌张力正常,注意力不集中,多动,冲动,重复刻板行为(反复摆弄纸、笔,撤去纸笔后反应激烈),言语延迟。化验检查:血常规、肝、肾功能、血电解质未见异常。生长激素激发试验峰值9.33 ng/mL,甲状腺功能示促甲状腺激素(TSH)7.235 μIU/mL。肾上腺皮质功能示促肾上腺皮质激素(ACTH)21.5 pg/mL,皮质醇(Cor)28.4 μg/dL。25羟维生素D 27.2 nmol/L。血氨基酸及酰基肉碱检测未见明显异常,尿液有机酸综合检测未见明显异常。心脏彩超:三尖瓣轻度反流,左室收缩、舒张功能正常。甲状腺B 超示未见明显异常。肾上腺B 超:双侧肾上腺区未见明显肿物。腹部B 超:肝胆胰脾及双肾未见明显异常。颅脑磁共振成像(MRI):脑白质斑点状异常信号(考虑脱髓鞘斑?),部分副鼻窦炎性改变。垂体MRI 示垂体形态正常,高度6 mm,信号未见异常,垂体柄居中,无增粗,视交叉无移位。韦氏儿童智力量表测试示语言智商(VIQ)79,操作智商(PIQ)68,全量表智商(FIQ)73,属于临界范围。
遗传学检查:患儿于外院完善染色体微阵列检查,对核基因组进行拷贝数变异分析结果显示其染色体17p11.2 区域存在约3.7 Mb 的杂合重复,该区域包含主要的功能基因RAI1、FLCN、ALDH3A2等,该区域拷贝数变异(CNVs)与Potocki-Lupski 综合征相关。结合患儿临床特点及染色体微阵列检测结果,PTLS 诊断明确。本例患儿的重复片段中还包含与努南综合征发病相关的PTPN11、SOS1、SHOC2、CBL 基因[3]。
治疗:家属要求改善患儿身高的意愿强烈,查阅国内外文献,有应用rhGH 治疗PTLS 患者的先例,并未报道治疗后出现不良反应[4],遂予患儿rhGH 0.13 IU/(kg·d)皮下注射,维生素D800 IU/d,左旋甲状腺素片12.5 μg/d,并嘱其针对自闭症行为进行行为干预,患儿于我院门诊定期随诊,现已治疗1 年9个月,患儿目前7 岁11 个月,身高121.5 cm(图1),由治疗前的-2.58 SD 增长至-1.23 SD,平均年身高增长速率为8.86 cm/年。治疗期间检测血常规、肝肾功能、甲状腺功能、空腹血糖胰岛素均正常,胰岛素样生长因子(IGF)-1 及胰岛素样生长因子结合蛋白3(IGF-BP3)均在正常范围内,复查肾上腺皮质功能正常,25 羟维生素D 较前上升(随诊结果见表1),复查头颅MRI 示双侧大脑半球、脑干及小脑形态、信号均未见明显异常。复查垂体MRI 示垂体形态正常,高度5.6 mm,信号未见异常,垂体柄居中,无增粗。监测心脏彩超示三尖瓣轻度反流,左室收缩、舒张功能正常。未出现脊柱侧弯。韦氏儿童智力量表测试示VIQ=55,PIQ=58,FIQ=55,属于智力低下范围。患儿6 岁7 个月(rhGH 治疗6 个月)监测睾丸B超示左侧精索鞘膜积液,于外院行精索鞘膜积液手术,术后未见不良反应。

表1 患儿接受rhGH 治疗随诊表
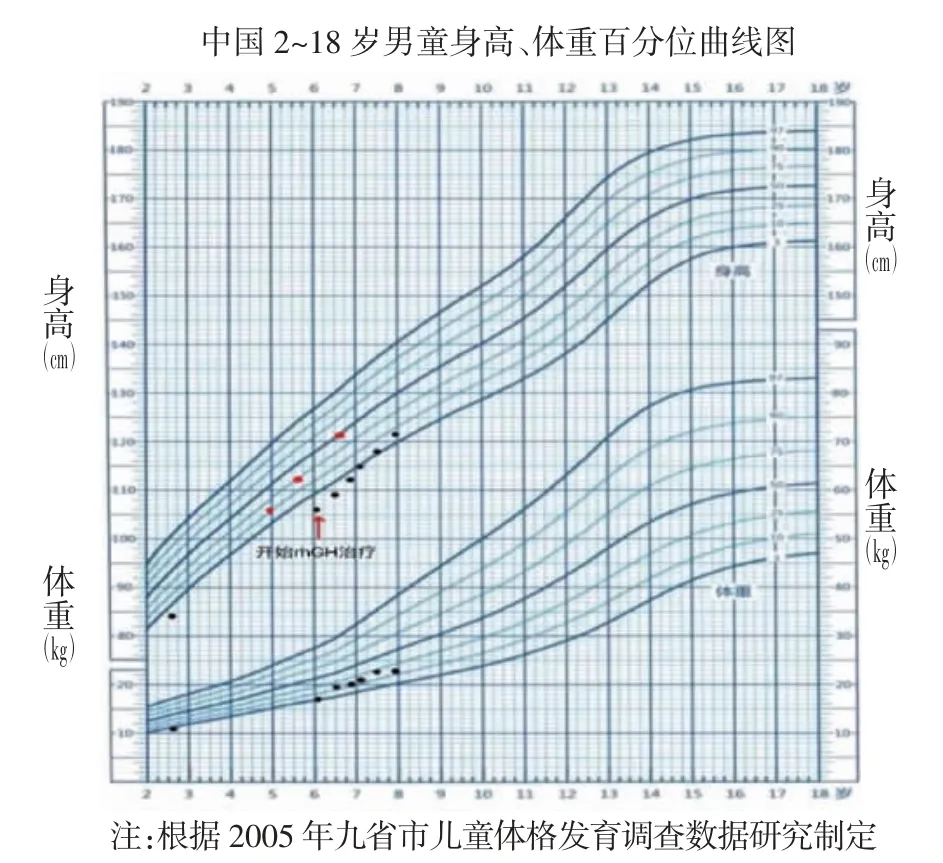
图1 患儿身高、体重百分位曲线图[5]
2 讨论
Potocki-Lupski 综合征是临床上罕见的染色体病,是由于包含RAI1(retinoic acid induced 1,维甲酸诱导1)在内的17p11.2 微重复所致。临床特点包括以发育迟缓、婴儿期肌张力低下、吞咽困难导致的喂养困难、睡眠呼吸暂停、结构性心血管异常、智力障碍、行为异常(包括注意力不集中、多动、沟通障碍、重复刻板行为、自闭症)、身材矮小、生长激素缺乏相关性低血糖及轻微的面部畸形(三角脸、宽额头、高腭弓)等[1-2,6]。另外,畸形足、肾脏异常、远视、斜视、轻度高频感神经听力损失、错颌畸形、肾上腺皮质功能不全、脑白质异常等不常见表现也有报道。染色体微阵列分析技术(chromosome microarray analysis,CMA)是本病的首选检测方法[7]。
尽管在17p11.2 微重复的序列范围内有许多基因,但有文献报道,导致PTLS 发生的的最小的重叠区域长度为125 kb,这其中仅包括RAI1,证明了RAI1 在PTLS 的表型中起关键作用[8]。动物实验也佐证了RAI1 是该表型的剂量敏感基因[9]。RAI1在许多组织中都有表达,它编码的转录因子直接参与神经元发育与神经分化、突触传递,这与PTLS 的行为异常表型相关[2,9-10],该基因还与细胞生长和细胞周期调节、骨骼和骨骼发育、脂质和葡萄糖代谢、行为功能和昼夜活动有关[2,11]。另外,FLCN 编码滤泡素,在肾脏中高度表达。PTLS 的肾脏表型与小鼠模型均表明FLCN 可能是肾脏异常的一个潜在因素[11]。
有文献报道,37 例PTLS 患者有9 例存在身材矮小。其中有2 例患者确诊存在生长激素缺乏症(growth hormone deficiency,GHD),1 例患者垂体MRI 显示脑垂体存在异常(垂体体积小、垂体后叶组织异位、垂体柄缺失),这3 例患者以及另外2 例身材矮小但未诊断GHD 的患者进行了生长激素治疗,身高均从中获益[4],但这5 例患者均未报道存在努南综合征(Noonan syndrome,NS)相关基因。本例患者应用生长激素治疗1 年9 个月,身高共增长1.35 SD,平均年身高增长速率为8.86 cm/年,提示治疗有效。因此对于矮小的PTLS 患者,可以适当考虑予以rhGH 治疗。
由于本例患者的重复片段中还包含PTPN11、SOS1、SHOC2、CBL 基因,这些基因与NS 的发病相关,NS 的常见临床表现包括特殊面容、先天性心脏病、身材矮小、胸廓畸形、视力异常等[3]。目前,关于17p11,2 微重复区域内包含NS 相关基因罕有报道,由于NS 与PTLS 的部分临床表型相重叠,这可能会导致重叠临床表型(身材矮小、鸡胸、远视、特殊面容、智力障碍等)的加重。国外有文献报道,PTLS 患儿的语言能力会随年龄增长及言语治疗改善[1],但本例患儿智力测试趋势显示与经典PTLS 患儿不同,这可能与其重复区域内包含NS 相关基因,从而使得智力受损有关。
综上所述,本文报道了1 例以身材矮小伴智力落后就诊的患儿,患儿存在匀称性身材矮小、远视、鸡胸、特殊面容、自闭症谱系障碍等表现。染色体微阵列检查提示存在包含RAI1 在内的17p11.2 区域3.7 Mb 微重复,诊断PTLS,同时重复区域内还包含PTPN11 等NS 相关基因,这可能导致两种综合征中重叠表型的加重。此外,rhGH 可作为改善PTLS 合并NS 的患儿身高的治疗方法,但由于目前没有大样本量及足够疗程的随访资料,其安全性及有效性还有待进一步的研究。
猜你喜欢
今日农业(2022年1期)2022-06-01 06:18:04
天津医科大学学报(2021年1期)2021-01-26 00:57:22
昆明医科大学学报(2020年12期)2021-01-26 00:44:02
中国生殖健康(2020年6期)2020-02-01 06:29:00
小雪花·初中高分作文(2019年8期)2019-10-07 08:46:42
新医学(2019年4期)2019-09-10 07:22:44
中国生殖健康(2018年6期)2018-11-06 07:09:38
海南医学(2016年8期)2016-06-08 05:43:00
磁共振成像(2015年9期)2015-12-26 07:20:34
中国医疗美容(2015年4期)2015-04-27 02:24:00
