
Altered gut microbiota composition in children and their caregivers infected with the SARS-CoV-2 Omicron variant
2023-06-25 08:54:18YiZhongWangJianGuoZhouYanMingLuHuiHuFangFeiXiaoTingGeXingWangLuZhengLianHuYuJunLeHuiYuGuangJunYuQiangXiaTingZhangWenHaoZhou
World Journal of Pediatrics 2023年5期
Yi-Zhong Wang · Jian-Guo Zhou · Yan-Ming Lu · Hui Hu · Fang-Fei Xiao · Ting Ge · Xing Wang · Lu Zheng · Lian-Hu Yu · Jun Le · Hui Yu · Guang-Jun Yu · Qiang Xia · Ting Zhang · Wen-Hao Zhou
Abstract Background Gut microbiota alterations have been implicated in the pathogenesis of coronavirus disease 2019 (COVID-19).This study aimed to explore gut microbiota changes in a prospective cohort of COVID-19 children and their asymptomatic caregivers infected with the severe acute respiratory syndrome coronavirus type 2 (SARS-CoV-2) Omicron variant.Methods A total of 186 participants,including 59 COVID-19 children,50 asymptomatic adult caregivers,52 healthy children (HC),and 25 healthy adults (HA),were recruited between 15 April and 31 May 2022.The gut microbiota composition was determined by 16S rRNA gene sequencing in fecal samples collected from the participants.Gut microbiota functional profiling was performed by using Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt) software.Results The gut microbiota analysis of beta diversity revealed that the fecal microbial community of COVID-19 children remained far distantly related to HC.The relative abundances of the phyla Actinobacteria and Firmicutes were decreased,whereas Bacteroidetes,Proteobacteria,and Verrucomicrobiota were increased in COVID-19 children.Feces from COVID-19 children exhibited notably lower abundances of the genera Blautia,Bifidobacterium,Fusicatenibacter,Streptococcus,and Romboutsia and higher abundances of the genera Prevotella,Lachnoclostridium,Escherichia-Shigella,and Bacteroides than those from HC.The enterotype distributions of COVID-19 children were characterized by a high prevalence of enterotype Bacteroides.Similar changes in gut microbiota compositions were observed in asymptomatic caregivers.Furthermore,the microbial metabolic activities of KEGG (Kyoto Encyclopedia of Genes and Genomes) and COG (cluster of orthologous groups of proteins) pathways were perturbed in feces from subjects infected with the SARS-CoV-2 Omicron variant.Conclusion Our data reveal altered gut microbiota compositions in both COVID-19 children and their asymptomatic caregivers infected with the SARS-CoV-2 Omicron variant,which further implicates the critical role of gut microbiota in COVID-19 pathogenesis.
Keywords Caregivers · Children · Coronavirus disease 2019 (COVID-19) · Gut microbiota · Severe acute respiratory syndrome coronavirus type 2 (SARS-CoV-2)
Introduction
The coronavirus disease 2019 (COVID-19) pandemic caused by severe acute respiratory syndrome coronavirus 2 (SARSCoV-2) has wreaked havoc on global health,and it is still ongoing due to continuously emerging variants of concern (VOC) [1].The recently emerged VOC Omicron (B.1.1.529) has rapidly spread worldwide and become the dominant variant [2].Clinical evidence has shown that Omicron causes symptoms similar to those of previously noted variants and tends to be more frequent in younger populations [3,4].Although the risks of hospitalization and fatality were lower in persons infected with Omicron than the original SARS-CoV-2 (index strain) and earlier prevalent variants,Omicron exacerbated the existing public health threats due to its notably increased transmission rate [5].Studies have suggested that strong binding to the entry receptor,angiotensin-converting enzyme 2 (ACE2) [6],and the ability to evade antibodies induced by previous infection or vaccination may be responsible for its high prevalence [7,8].
Many studies have indicated the involvement of the gastrointestinal (GI) tract,suggesting the manifestation of GI disorders/symptoms during COVID-19 [9–11].Studies have also revealed that rectal swabs are persistently positive in some children,even nasopharyngeal testing is negative,and the virus has the capability to replicate in enterocytes [10,12].A considerable proportion of patients with COVID-19 develop at least one GI symptom,such as abdominal pain,nausea or vomiting,and diarrhea [11].Certain studies have reported altered gut microbiota composition in patients with COVID-19 and implicated its potential role in COVID-19 pathogenesis [13,14].However,gut microbiota profiling in children with COVID-19 is still limited,and the impact of Omicron on host microbiota composition remains to be determined.In late February 2022,during the Omicron wave in Shanghai,China,the sub-lineage BA.2.2 was detected and confirmed through phylogenetic analysis in 129 patients.Since March 2022,approximately 3500 reported pediatric cases were admitted to the hospital in Shanghai (from April to May 2022) [15,16].Herein,we aimed to investigate the alterations in gut microbiota composition in a prospective cohort of pediatric COVID-19 patients and their asymptomatic caregivers infected with the SARSCoV-2 Omicron variant from a designated hospital during the wave of the Omicron variant BA.2 pandemic in Shanghai,China.
Methods
Study cohort
This prospective cohort study was conducted at Shanghai Children’s Hospital and Renji Hospital,School of Medicine,Shanghai Jiao Tong University.According to the local government policy,all pediatric COVID-19 patients were required to be admitted to a designated hospital,and adult family members were permitted to take care of their children in ward regardless of SARS-CoV-2 infection status.All COVID-19 patients were confirmed by a positive RT-PCR test for SARS-CoV-2 in nasopharyngeal swabs or nasal swabs.A total of 109 participants,including 59 COVID-19 children (below 16 years old) and 50 asymptomatic caregivers (below 60 years old),were recruited for this study between 15 April and 31 May 2022.COVID-19 patients with underlying diseases were excluded from this study.Data regarding demographics,clinical symptoms,and disease duration were recorded and analyzed.
Fifty-two healthy children (HC) and 25 healthy adults (HA) age-and gender-matched to COVID-19 children and asymptomatic caregivers were recruited as healthy controls.Each healthy individual was SARS-CoV-2 negative,confirmed by RT-PCR test for SARS-CoV-2 in nasopharyngeal swabs or nasal swabs,and had no history of SARSCoV-2 infection.None of the healthy controls showed relevant COVID-19 or signs of symptoms at the time of fecal sample collection.Additionally,the people who had probiotics or antibiotic therapy within six months and presented symptoms of any infection were considered under the exclusion criteria during the recruitment of healthy controls.A total of 186 fecal samples were collected for gut microbiome analysis.Each participant provided a single fecal sample.All fecal samples were collected using sterile containers and stored at -80 °C until DNA extraction.
The study was carried out in compliance with the Helsinki Declaration (as revised in 2013) and was approved by the Regional Ethical Review Board of Shanghai Children’s Hospital (2022R071-E01).Informed consent to participate in the study was obtained from participants (or their parent or legal guardian in the case of children under 16).
Stool DNA extraction,PCR amplification,and sequencing
Stool DNA was extracted from feces by using a Stool DNA Kit (D4015-02,Omega,USA) according to the manufacturer’s instructions.16S rRNA gene fragments (V3-V4) were amplified by using primers 338F (5′-ACT CCT ACG GGA GGC AGC AG-3′) and 806R (5′-GGA CTA CHVGGG TWT CTAAT-3′).PCR amplification was performed using a TransStart FastPfu DNA Polymerase Kit (TransGen Biotech,Beijing,China) with 10 ng of template DNA and 0.5 μM forward/reverse primers.The thermocycling conditions for amplification were as follows:1 minute at 95 °C,30 seconds at 95 °C,30 seconds at 55 °C,and 45 seconds at 72 °C for 30 cycles,and an extension of 10 minutes at 72 °C.The PCR products were purified using the AxyPrep DNA Gel Extraction Kit (Axygen Biosciences,Union City,CA,USA).Sequencing libraries generated using a TruSeqR DNA PCRFree Sample Preparation Kit (Illumina,USA) from purified amplicons were subjected to paired-end sequencing on an Illumina MiSeq PE300 platform (Illumina,Inc.,CA,USA).
16S rRNA gene sequence analysis
The raw sequencing data were quality filtered by fastp and were merged by FLASH (v1.2.11).Operational taxonomic units (OTUs) were generated from denoised high-quality sequences by using DADA2 after removing chimeras in the unique sequences [17].Taxonomic assignment of OTUs was conducted using the Blast consensus taxonomy classifier implemented in QIIME2 and the SILVA 16S rRNA database (v138).The alpha diversity was measured using the Shannon,Simpson,Chao1,and Observed species,and the beta diversity among samples was calculated through principal coordinate analysis (PCoA) with the Bray–Curtis distance and Jaccard,unweighted and weighted UniFrac metrics based on the relative OTU abundances.Permutational multivariate analysis of variance analysis was used to assess the significant differences in the gut microbiome structure between the two groups.The significant differences in alpha diversity and the relative abundances of bacterial taxa were assessed by Wilcoxon rank-sum test between two groups withPvalues adjusted for false discovery rate.The Dirichlet multinomial mixtures (DMM) approach on a genus-level matrix of samples from all participants was performed to stratify the joint microbiome dataset into enterotypes as described by Holmes et al.and applied in R [18].The observed genus richness was calculated on the relative microbiome profiling matrix (downsized to 10,000 reads) using phyloseq and only considered the core taxa that were prevalent above 0.1% relative abundance in 50% of the samples.Bacterial taxa enrichments were analyzed by linear discriminant analysis (LDA) effect size (LEfSe).
Functional profiling
Gut microbiota functional profiling was performed using phylogenetic investigation of communities by reconstruction of unobserved states software [19].Functional metabolic pathway enrichment analysis was performed using Kyoto Encyclopedia of Genes and Genomes (KEGG) and the cluster of orthologous groups of proteins (COG) database based on the predicted metagenome.LEfSe was used to detect the significant changes in predicted KEGG and COG pathways between groups with a significanceα-value (on factorial Kruskal–Wallis test) of 0.01 and an effect LDA score of 3.0.
Statistical analysis
Data are presented as numbers (percentage) for dichotomous variables and medians with interquartile ranges (IQRs,25–75th percentiles) for continuous variables.The Chi-squared test was used to analyze the distribution of dichotomous variables.The continuous variables between two groups were compared by the Wilcoxon rank-sum test.All statistical analyses were conducted by using SPSS software (version 20.0),and statistical significance was defined as aPvalue < 0.05.
Results
Characteristics of the study cohort
As shown in Table 1,the median age of the 59 pediatric COVID-19 patients was 4.8 years (IQR=1.8–7.9 years),and 29 (49.2%) were boys.The mode of delivery was comparablebetween the COVID-19 children and HC.The COVID-19 vaccination rate of COVID-19 children was 25.4% (15/59).All pediatric patients were classified as either mild (n=45,76.3%) or moderate (n=14,23.7%) conditions,and fever was the most common symptom (n=48,81.4%),followed by cough (n=27,45.8%).GI symptoms of abdominal pain,diarrhea,and vomiting were observed in 1 (1.7%),2 (3.4%),and 2 (3.4%) pediatric patients,respectively.The median duration of diagnosis to recovery of the COVID-19 children was 12 days (IQR=10–14.5 days).All recruited adult caregivers were asymptomatic carriers with an RT-PCR confirmation of SARS-CoV-2 infection,and the majority of the caregivers were parents of pediatric patients.There was no significant difference in demographic characteristics,including age,sex,and COVID-19 vaccination rate,between COVID-19 children and adult caregivers and their corresponding healthy controls.
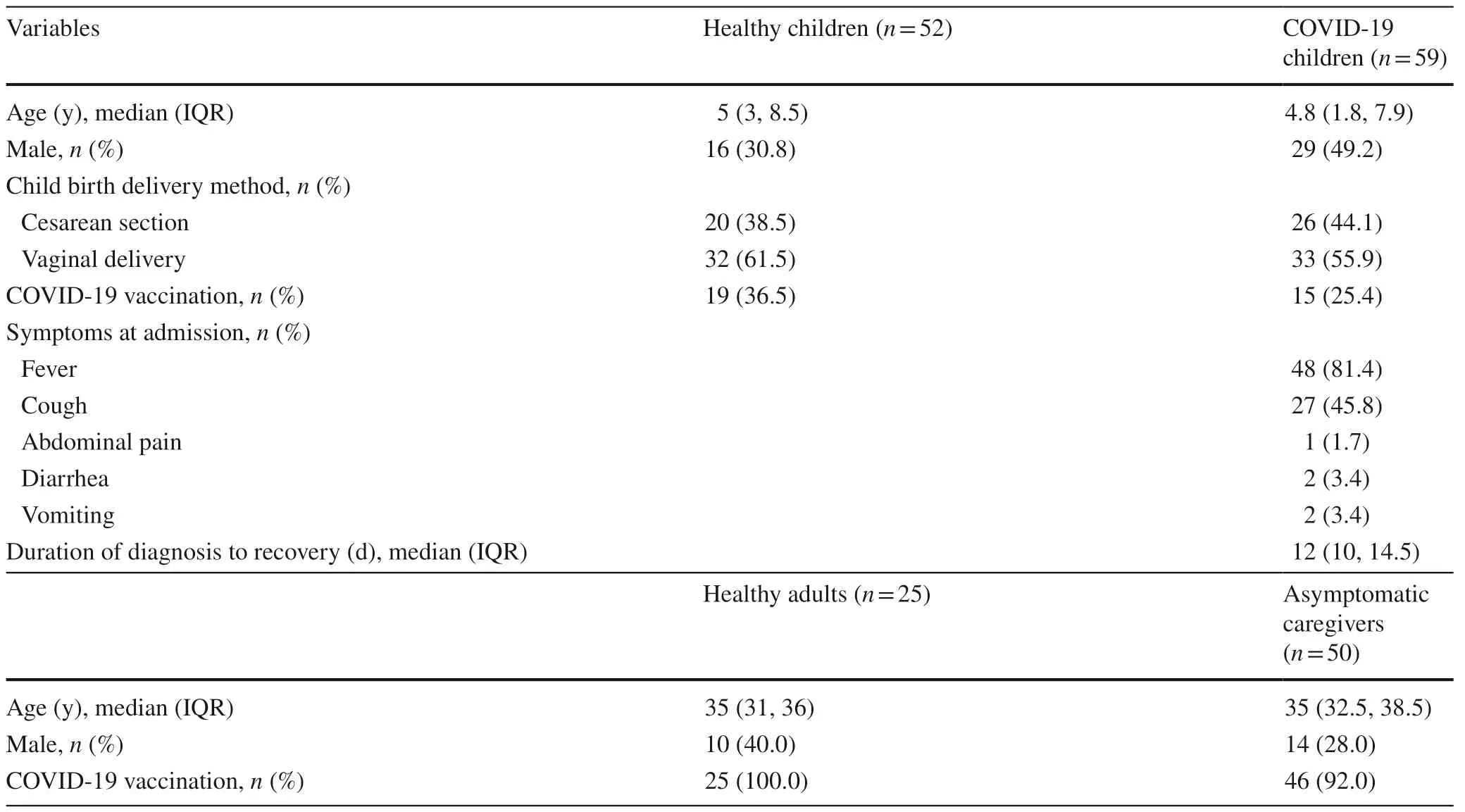
Table 1 Demographic and clinical characteristics of the study cohort
SARS-CoV-2 Omicron infection induces gut microbiota composition alteration
To characterize the gut microbiota composition in children infected with the SARS-CoV-2 Omicron variant,bacterial 16S rRNA gene sequencing was performed in fecal samples from 59 COVID-19 children and 52 HC.After sequence processing and filtering,the average read count per sample was 75,158,ranging from 42,439 to 120,103.No signifi-cant difference in the alpha diversity of bacterial richness was observed between COVID-19 children and HC,except for the Simpson index (Supplementary Fig.1a).The PCoA profile of microbial beta diversity using Bray–Curtis distance and Jaccard revealed significant clustering of samples according to the occurrence of COVID-19 (Fig.1 a).In contrast with the HC,the abundances of the dominant bacterial phylaActinobacteriaandFirmicuteswere significantly decreased,whereas the abundances of the phylaBacteroidetes,Proteobacteria,andVerrucomicrobiotawere increased in COVID-19 children (Supplementary Fig.2a,b).The dominant genera wereBacteroides,Bifidobacterium,Blautia,andFaecalibacteriumin both HC and COVID-19 children (Fig.1 c).To compare the taxonomic profiles between COVID-19 and HC,genera with a relative abundance larger than 0.01% in each sample were selected.The data showed that fecal samples from COVID-19 children exhibited higher relative abundances ofFusobacterium,Akkermansia,Sutterella,Veillonella,Lachnospira,Parasutterella,Prevotella,Parabacteroides,Lachnoclostridium,Escherichia-Shigella,andBacteroidesand lower relative abundances ofCollinsella,Eubacterium_hallii_group,Dorea,Fusicatenibacter,Romboutsia,Streptococcus,Blautia,andBifidobacteriumthan those of HC (Fig.1 d).
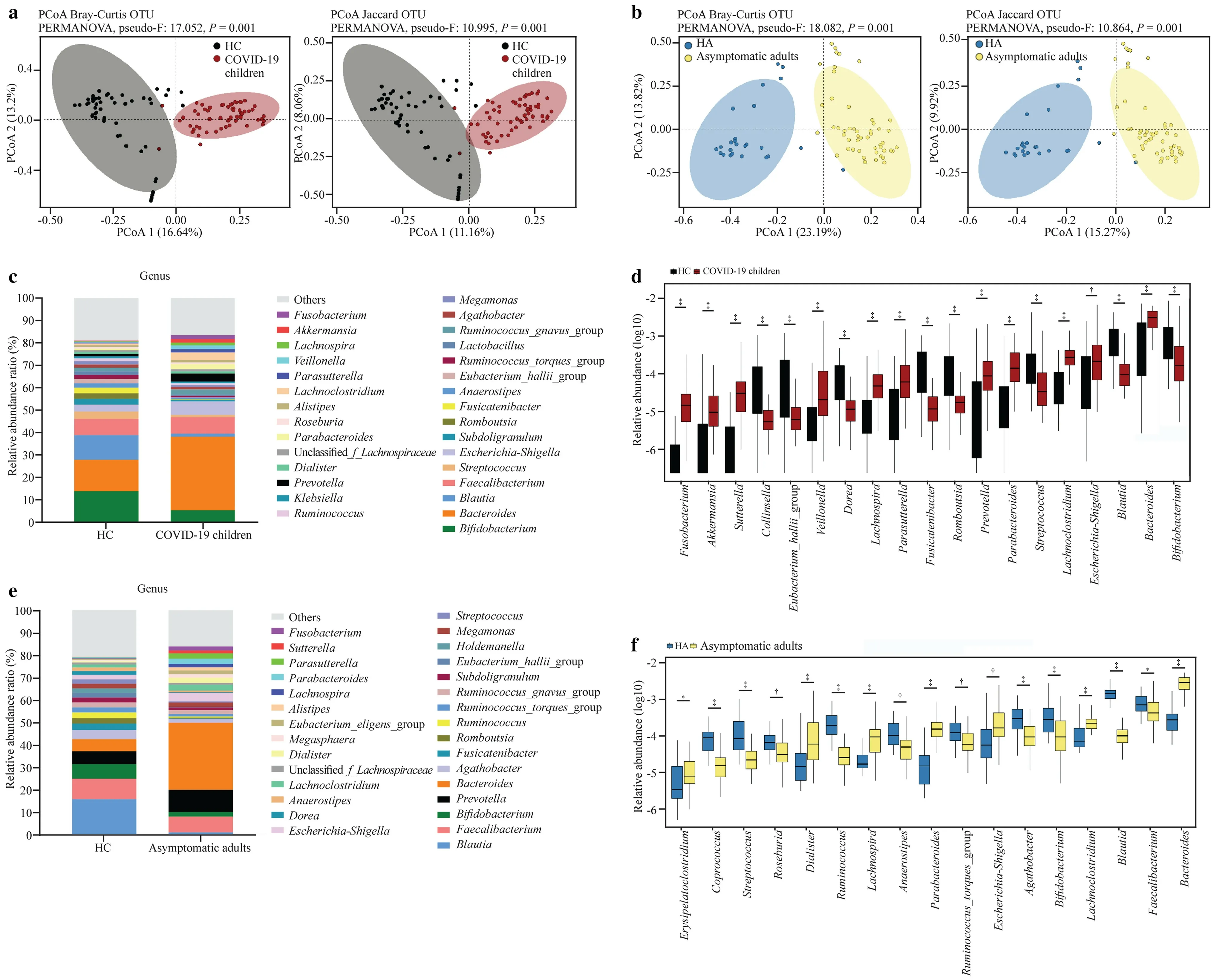
Fig.1 Altered gut microbiota diversity and compositions of COVID-19 children and asymptomatic caregivers in comparison with healthy children (HC) and healthy adults (HA).a,b Beta diversity analyzed by PCoA of Bray–Curtis distance and Jaccard between groups.Each dot represents one sample,and the ellipses represent groups.Groups were compared by using the PERMANOVA method; c,e relative abundance ratio of bacteria at the genus level in different groups.The top 30 abundant genera are indicated; d,f genera with signifi-cant differences between groups.The relative abundances are shown in log10.Statistical significance was assessed by using the Wilcoxon rank-sum test,with *P ≤ 0.05,† P ≤ 0.01,‡ P ≤ 0.001,§ P ≤ 0.0001.COVID-19 2019,PCoA principal coordinate analysis,OTU operational taxonomic units,PERMANOVA permutational multivariate analysis of variance
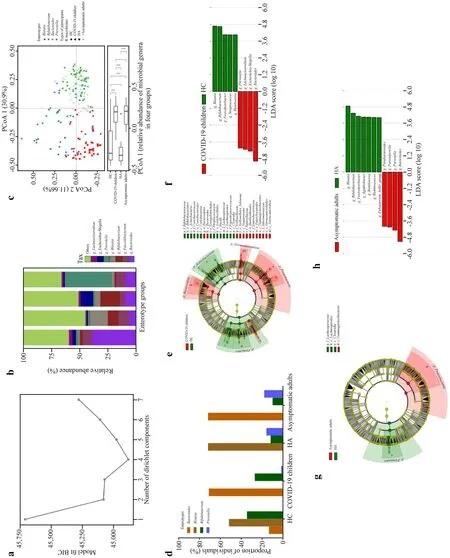
Fig.2 Bacterial features associated with SARS-CoV-2 Omicron infection.a Information criteria from the DMM community typing of the joint microbiome dataset of all subjects (186 samples) showing the optimum (minimum BIC) at four Dirichlet components; b the barplot showing the four enterotype groups identified by DMM,with average relative abundance of the representative genera in each enterotype group; c principal coordinate analysis visualization of interindividual differences (Bray–Curtis distance) in relative microbiome profiles in each group (Wilcoxon rank-sum test,with § P ≤ 0.0001); d barplot showing the enterotype prevalence distributions in each group; e,g cladogram generated from LEfSe analysis showing the relationship between taxa (from the inner ring to the outer ring,the grades represent the phylum,class,order,family,genus,and species).Each dot represents a taxonomic hierarchy; f,h LEfSe identified the taxa with the greatest differences in abundance between the two groups.Bacteria with an LDA score > 3 were plotted.DMM Dirichlet multinomial mixtures,BIC Bayesian information criterium,LDA linear discriminant analysis,HC healthy children,HA healthy adults,COVID-19 coronavirus disease 2019,SARS-CoV-2 severe acute respiratory syndrome coronavirus 2
Regarding the gut microbiota composition of asymptomatic caregivers,16S rRNA gene data were obtained from 50 asymptomatic caregivers and 25 HA.Similar to the results of the pediatric cohort,there was no significant difference in microbial alpha diversity (Supplementary Fig.1b),significant clustering of samples by PCoA profile of beta diversity (Fig.1 b),or taxonomic profile changes at the phylum (Supplementary Fig.2c,d) and genus levels (Fig.1 e,f) between asymptomatic caregivers and HA.
Information criteria from the DMM community typing of the COVID-19 children,HC,asymptomatic caregivers and HA showed the optimum (minimum Bayesian information criterium) at four Dirichlet components (n=186 biologically independent samples,Fig.2 a).The four enterotypes identified by DMM community typing on the joint microbiome dataset were labeledBacteroides,Blautia,Bifidobacterium,andPrevotellabased on enterotype-discriminating predominant genera (Fig.2 b).Each group displayed diverging enterotype prevalence distributions,and inter-individual variations in microbiome composition at the genus level were visualized by PCoA using Bray–Curtis dissimilarity (Fig.2 c).The enterotype prevalence distributions of SARSCoV-2-infected subjects were significantly different from those of healthy controls (Fig.2 c).As shown in Fig.2 d,the enterotype distribution of SARS-CoV-2-infected subjects was characterized by a high prevalence of enterotypeBacteroidesin both COVID-19 children (71%) and asymptomatic adults (72%),while healthy controls presented with a high prevalence of enterotypeBlautia(52% in HC,72% in HA).In addition,a higher prevalence of the enterotypeBifidobacteriumwas observed in children (34% in HC,27% in COVID-19 children) than in adults (12% in HA,10% in asymptomatic adults),and the enterotypePrevotellawas more prevalent in adults (0% in HC,2% in COVID-19 children;16% in HA,18% in asymptomatic adults).
Furthermore,LEfSe analysis identified multiple taxa at different levels that were differentially abundant in the COVID-19 children and HC (Fig.2 e),asymptomatic caregivers and HA (Fig.2 g),respectively.Specifically,LEfSe feature selection with an LDA score > 3.0 identified several bacterial genera that discriminated the subjects infected with the Omicron variant and HC,including notably higher abundances ofBlautia,Bifidobacterium,Fusicatenibacter,Streptococcus,andRomboutsiain HC,and enrichedPrevotella,Lachnoclostridium,Escherichia-Shigella,andBacteroidesin COVID-19 children (Fig.2 f).Similarly,Blautia,Bifidobacterium,Fusicatenibacter,Agathobacter,Romboutsia,Ruminococcus,andEubacterium_hallii_group were enriched in HA,andParabacteroides,Parasutterella,Prevotella,andBacteroideswere abundant in asymptomatic caregivers (Fig.2 h).
Functional profile analysis of the gut microbiota
KEGG gene and genomic profiling and COG metabolomic profiling did not provide a significant difference at large.The relevant functional pathways mentioned along with mean proportion values are detailed in Fig.3 and Supplementary Tables 1 and 2.The mean proportion observations showing values more than < 0.5% are presented in graphical form (Fig.3),and values less than < 0.5% are detailed in Supplementary Table 1 (for KEGG genomic profiling) and Supplementary Table 2 (for COG metabolomic profiling).The results reported by KEGG profiling in COVID-19 children appeared to show more activity in glycan biosynthesis and metabolism (2.07% mean proportion) and molecular signaling interactions (6.38% mean proportion) than HC,with values of 1.59% and 5.51%,respectively.On the other hand,the asymptomatic adults were reported to have higher functional activity through KEGG,as the cellular community (2.82%) and sensory system (9.67%) compared to HA had values of 1.06% and 2.51%,respectively.The membrane transport and molecular signaling and interaction pathways revealed values of 2.82% and 3.19%,respectively.However,the mean proportion values less than HA were 3.94% and 5.57%,respectively.
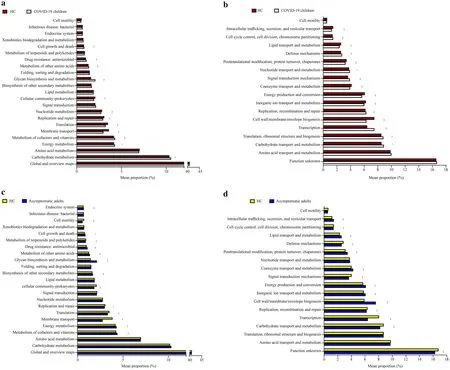
Fig.3 Functional divergence between the gut microbiota of SARSCoV-2 Omicron-infected subjects and healthy controls.The bar charts showing KEGG on level 2 (a,c) and COG (b,d) pathways with significant differences.Functional pathways with a mean proportion larger than 0.5% in both groups are shown.Comparisons were performed between COVID-19 children and healthy children (HC),asymptomatic caregivers and healthy adults (HA) at KEGG pathway level 2 and COG pathway,respectively.Significance was determined by using Welch’s t test with P values adjusted by the Benjamini–Hochberg correction procedure for controlling the false discovery rate,*P ≤ 0.05,†P ≤ 0.01,‡P ≤ 0.001,§ P ≤ 0.0001.COVID-19 coronavirus disease 2019,SARS-CoV-2 severe acute respiratory syndrome coronavirus 2,KEGG Kyoto Encyclopedia of Genes and Genomes,COG cluster of orthologous groups of proteins
COG functional metabolic profiling is represented in Supplementary Table 2 and Fig.3 b,d.Cell wall/cell membrane/envelop biogenesis and energy production and conversion in COVID-19 children were increased in mean proportions of 7.48% and 6.07%,respectively,but transportation metabolism was decreased by 6.4%.In the case of HC,the cell wall/cell membrane and envelop biogenesis functional activity decreased by 6.31% and 5.68%,respectively,but the transportation functional activity increased by 7.48%.Similarly,for asymptomatic adults,the cell wall/membrane/envelop biogenesis activity and energy production and conservation activity were reported to be 7.59% and 6.13%,respectively,compared to HA,which were 5.93% and 5.73%,respectively.However,transportation functionality is reduced in asymptomatic adults,such as COVID-19 children.However,this activity was increased in HA.
Discussion
The global COVID-19 pandemic is currently ongoing due to the emergence of SARS-CoV-2 VOC,such as the Omicron variant [2].In March 2022,a tsunami of SARS-CoV-2 Omicron infection attacked Shanghai,one of the largest cities in China,which affected a population greater than 600,000 people,including more than 12,000 children,in a short period of time [15,16].Consistent with previous reports [5],Omicron infection caused less severe disease,as the majority of the cases were asymptomatic carriers in Shanghai [15,16].In this study,all enrolled pediatric COVID-19 patients were mild or moderate cases with common symptoms of fever and cough and recovered in a median duration of 10 days,and the recruited adult caregivers were asymptomatic carriers with a PCR confirmation of SARS-CoV-2 infection.
The resident microbiota,consisting of trillions of microbes and their derived metabolites in the GI tract,are critical regulators in modulating host immune defense against pathogen infections [20].It has been shown that respiratory infections are associated with gut microbiota dysbiosis in children [21].Enterococcuswas increased,whereas the abundances ofEubacterium,Faecalibacterium,andBifidobacteriumwere reduced in children with recurrent respiratory tract infections [22].An expansion of the familiesClostridiales,Odoribacteraceae,Lactobacillacea,andActinomcyceswas observed in respiratory syncytial virus-infected children compared to healthy controls,and the enriched genusBacteroideswas positively associated with bronchiolitis severity [23,24].Children with pulmonary tuberculosis had enrichedPrevotellaandEnterococcusand decreasedRuminococcaceae,Bifidobacteriaceae,andFaecalibacterium prausnitziicompared to their healthy controls [25].DecreasedBifidobacteriawas found in the intestinal flora of children withMycoplasma pneumoniaepneumonia [26].Changed gut microbiota compositions in SARS-CoV-2-infected subjects and their associations with COVID-19 progression have been reported in previous studies [13,14].The disturbed gut microbiota in adult COVID-19 patients was characterized by a decreased relative abundance of commensal/beneficial bacteria that produce an anti-inflammatory response (Lachnospira,Roseburia,Eubacterium,andFaecalibacterium prausnitzii) and increased relative abundances of opportunistic pathogens (Clostridium hathewayi,Enterobacteriaceae,Enterococcus,Actinomyces viscosus,andBacteroides nordii) [13,27].There have been limited studies investigating the gut microbiota composition in children infected with SARS-CoV-2.Enrichments ofPseudomonas,Herbaspirillum,andBurkholderiawere observed in children during the early stages of COVID-19 [28].The bacteria linked to anti-inflammation,such asBifidobacterium bifidumandAkkermansia muciniphila,were decreased in asymptomatic very young children with SARS-CoV-2 infection [29].A recent study showed that the relative abundance ofFaecalibacterium prausnitziiwas reduced,whereas that ofEggerthella lentawas increased in COVID-19 children [30].
To our knowledge,this study is the first to show the gut microbiota composition of COVID-19 children and their caregivers with SARS-CoV-2 Omicron variant infection.Consistently,our data revealed that the microbial diversity and composition in the Omicron-infected children and their caregivers differed from those of healthy controls,such as notably higher abundances of the generaEscherichia-Shigella,Parabacteroides,Parasutterella,Prevotella,andSutterellaand lower relative abundances ofBifidobacterium,Blautia,Collinsella,andDorea.We further observed four enterotypes (Bacteroides,Blautia,Bifidobacterium,andPrevotella) by using DMM community typing on the joint microbiome dataset.The results revealed that the enterotype distributions of SARS-CoV-2-infected subjects were significantly different from those of healthy subjects,which was characterized by a high prevalence of enterotypeBacteroidesin both COVID-19 children and asymptomatic adults.EnrichedBacteroidesandPrevotellaabundances in fecal samples from individuals with SARS-CoV-2 infection in both COVID-19 children and asymptomatic adults may be used as a marker of infection,and increasedLachnoclostridiumandEscherichia_Shigellaobserved in COVID-19 children have the potential to be a marker of the presence of symptoms.Nevertheless,studies with large cohorts are needed to validate these findings.In addition to the direct virus/infection effect on microbiota composition,changes in lifestyle during the pandemic,such as mitigation strategies (community lockdown,school closure,etc.) might also affect the microbiota composition in individuals infected with SARS-CoV-2,as well as the healthy controls.Such effects need to be investigated in future studies.Given that the gut microbiota composition can be influenced by delivery mode,antibiotic use,nutritional preferences,pets,etc.,in infants and young children,these microbiota-related factors should be considered in gut microbiota studies [31].Unfortunately,we did not harmonize these factors for the microbiome analysis due to the lack of information and limited number of enrolled participants.
Currently,the exact mechanism of gut microbiota in the pathogenesis of COVID-19 remains unclear.Studies have shown that the altered gut microbiota composition is associated with disease severity concordant with elevated concentrations of inflammatory cytokines in patients with COVID-19,which suggests that the gut microbiota may be involved in COVID-19 progression by inducing cytokine storms [14].It was shown that enrichment of opportunistic pathogens and loss of beneficial bacteria were associated with active SARS-CoV-2 GI infection in COVID-19 patients [32].Grenga et al.highlighted a possible link between gut microbiota alterations and fecal SARS-CoV-2 RNA levels and altered molecular functions,including enterocyte damage,enhanced intestinal permeability,and immune response activation,which may contribute to disease severity in COVID-19 patients with high SARS-CoV-2 RNA levels in the GI tract [33].In post-COVID-19 patients,respiratory dysfunction was associated with altered gut microbiota and persistently elevated lipopolysaccharide (LPS)-binding protein levels,which implicated that the altered gut microbiota may affect the COVID-19 long-term outcome through the gut–lung axis [34].In addition,gut microbiota-derived metabolites and components,such as LPS,deaminotyrosine,and short-chain fatty acids (SCFAs),may trigger either innate or adoptive immunity against SARS-CoV-2 infection [13].Recently,gut microbiota composition was reported to be associated with SARS-CoV-2 vaccine immunogenicity [35].Here,we showed that the microbial metabolic activities in both KEGG and COG pathways were perturbed in subjects infected with SARS-CoV-2,suggesting that the altered gut microbiota-induced dysregulation of basic metabolic processes may be involved in the pathogenesis of COVID-19.Taken together,uncovering the role of gut microbiota dysbiosis in the pathogenesis of COVID-19 could shed light on the exploration of microbiota-based therapeutic strategies to fight SARS-CoV-2 infection.
Several limitations existed in the current study.First,this was a single-center,pilot observational study with a small recruited cohort.Multicenter studies with large cohorts are necessary to investigate the altered gut microbiota in children with SARS-CoV-2 infection.Second,we only evaluated the bacterial composition changes in the feces of COVID-19 patients by 16S rRNA sequencing and not by shotgun metagenomic sequencing.The changes in other microbiota components,including viruses and fungi,need to be investigated in future studies.Third,the major mediators of host-microbiota interactions,such as SCFAs,bile acids,amino acids,organic acids,and fatty acids,were not determined in the current study.Finally,microbiota resident in other body sites,particularly in the respiratory tract,were not investigated in this study.
In summary,our data indicate that the gut microbiota compositions were significantly altered in both children and their asymptomatic caregivers infected with the SARSCoV-2 Omicron variant.Future investigations are needed to explore the critical role of gut microbiota in COVID-19 pathogenesis.
Supplementary InformationThe online version contains supplementary material available at https:// doi.org/ 10.1007/ s12519-022-00659-6.
Author contributionsWYZ and ZJG contributed equally to this study.WYZ contributed to data curation,formal analysis,funding acquisition,visualization,and writing of the original draft.ZJG contributed to data curation and visualization.LYM and YH contributed to methodology.HH,WX,ZL,YLH,and LJ contributed to investigation.XFF contributed to data curation,formal analysis,and software.GT contributed to formal analysis and investigation.YGJ and XQ contributed to project administration,resources,and validation.ZT contributed to conceptualization,formal analysis,funding acquisition,methodology,project administration,resources,supervision,and reviewing and editing.ZWH contributed to conceptualization,project administration,resources,supervision,and reviewing and editing.All authors approve the final version of the manuscript.
FundingThis work was supported by the grants from the National Natural Science Foundation of China (No.81870373),and the Natural Science Foundation of Shanghai (No.22ZR1451800).The funders had no role in study design,data collection,and analysis,decision to publish,or preparation of the manuscript.
Data availabilityRaw sequencing data have been deposited in NCBI SRA database under study accession number PRJNA859805.
Declarations
Ethical approvalThis study was approved by the Ethics Review Committee of Shanghai Children’s Hospital,School of Medicine,Shanghai Jiao Tong University (2022R071-E01),and carried out in accordance with the ethical guidelines of the Helsinki Declaration (as revised in 2013).Informed consents to participate in the study have been obtained from participants (or their parent or legal guardian in the case of children under 16).
Conflict of interestNo financial or non-financial benefits have been received or will be received from any party related directly or indirectly to the subject of this article.Author Wen-Hao Zhou is a member of Editorial Board forWorld Journal of Pediatrics.The paper was handled by the other Editor and has undergone rigorous peer review process.Author Wen-Hao Zhou was not involved in the journal's review of,or decisions related to this manuscript.The authors have no conflict of interest to declare.
 World Journal of Pediatrics2023年5期
World Journal of Pediatrics2023年5期
- World Journal of Pediatrics的其它文章
- Editors
- Information for Readers
- Instructions for Authors
- New epidemiological trends of respiratory syncytial virus bronchiolitis during COVID-19 pandemic
- Retroperitoneal kidney transplantation with liver and native kidney mobilization:a safe technique for pediatric recipients
- Impact of the COVID-19 kindergarten closure on overweight and obesity among 3-to 7-year-old children