
Sphingosine phosphate lyase insufficiency syndrome:a systematic review
2023-06-25 08:54ZahraPournasiriAbbasMadaniFatemehNazarpackJohnSayerZahraChavoshzadehFatemehNiliPaulinaTranJulieSabaMahnazJamee
World Journal of Pediatrics 2023年5期
Zahra Pournasiri · Abbas Madani · Fatemeh Nazarpack · John A. Sayer · Zahra Chavoshzadeh · Fatemeh Nili · Paulina Tran · Julie D. Saba · Mahnaz Jamee,
Abstract Background Sphingosine-1-phosphate lyase insufficiency syndrome (SPLIS) or nephrotic syndrome type-14 is caused by biallelic mutations in SGPL1.Here,we conducted a systematic review to delineate the characteristics of SPLIS patients.Methods A literature search was performed in PubMed,Web of Science,and Scopus databases,and eligible studies were included.For all patients,demographic,clinical,laboratory,and molecular data were collected and analyzed.Results Fifty-five SPLIS patients (54.9% male,45.1% female) were identified in 19 articles.Parental consanguinity and positive family history were reported in 70.9% and 52.7% of patients,respectively.Most patients (54.9%) primarily manifested within the first year of life,nearly half of whom survived,while all patients with a prenatal diagnosis of SPLIS (27.5%) died at a median [interquartile (IQR)] age of 2 (1.4–5.3) months (P =0.003).The most prevalent clinical feature was endocrinopathies,including primary adrenal insufficiency (PAI) (71.2%) and hypothyroidism (32.7%).Kidney disorders (42,80.8%) were mainly in the form of steroid-resistant nephrotic syndrome (SRNS) and progressed to end-stage kidney disease (ESKD) in 19 (36.5%) patients at a median (IQR) age of 6 (1.4–42.6) months.Among 30 different mutations in SGPL1,the most common was c.665G > A (p.Arg222Gln) in 11 (20%) patients.Twenty-six (49.1%) patients with available outcome were deceased at a median (IQR) age of 5 (1.5–30.5) months,mostly following ESKD (23%) or sepsis/septic shock (23%).Conclusion In patients with PAI and/or SRNS,SGPL1 should be added to diagnostic genetic panels,which can provide an earlier diagnosis of SPLIS and prevention of ESKD and other life-threatening complications.
Keywords Nephrotic syndrome type 14 · Immunodeficiency · Lymphopenia · Sphingosine-1-phosphate lyase 1 · Sphingosine-1-phosphate lyase insufficiency syndrome
Introduction
Sphingosine-1-phosphate lyase insufficiency syndrome (SPLIS),also referred to as nephrotic syndrome type 14 (NPHS14),is a newly recognized rare autosomal recessive disorder caused by biallelic defects inSGPL1.SGPL1encodes sphingosine-1-phosphate (S1P) lyase,and consistent with its ubiquitous expression,patients exhibit a heterogenous spectrum of manifestations including congenital nephrotic syndrome,endocrinopathies,skin disorders,immunodeficiency,and neurological abnormalities [1].S1P lyase irreversibly catalyzes the cleavage of S1P in the terminal step of the sphingolipid catabolic pathway,forming phosphoethanolamine and hexadecenal,a long chain aldehyde.As the guardian of the only exit point of sphingolipid metabolism,S1P lyase is considered the major modulator of cellular sphingolipid catabolism [2].This pathway normally has regulatory roles through amplification of signals mediated by signal transducer and activator of transcription 3 and nuclear factor-kappa B,prevention of histone deacetylase activity,and inhibition of apoptosis.Therefore,aberrant S1P signaling,caused by the accumulation of S1P,may disrupt transcription,inflammation,and carcinogenesis [3,4].Most of S1P’s functions are ascribed to its ability to act as a ligand for a family of G protein-coupled receptors,although other functions such as its impact on calcium homeostasis have not been linked to S1P receptor signaling to date [5].
Although the first description of S1P lyase enzyme activity was reported in 1969 [6],defective humanSGPL1was first identified in 2017 by two independent research groups.Lovric et al.[7] performed whole exome sequencing in seven families with a combination of steroid-resistant nephrotic syndrome (SRNS),ichthyosis/acanthosis,primary adrenal insufficiency (PAI),immunodeficiency,and neurological problems.The authors identified nine different diseasecausing variants inSGPL1,causing diminished SGPL1 protein and/or S1P lyase activity,and impaired degradation of long-chain sphingoid bases.They further investigated the impact of missense mutations in theDrosophila SGPL1orthologSply(shown previously to exhibit neuromuscular and reproductive abnormalities [8]) and found reduced viability and podocytopathy and lack of rescue in sphingolipid profile.Prasad et al.[9] demonstrated fourSGPL1biallelic pathogenic variants through next generation sequencing in five kindreds affected by PAI,SRNS,ichthyosis,neurological dysfunction,primary hypothyroidism,and cryptorchidism.They subsequently showed histological abnormalities in glomeruli and adrenals ofSgpl1knockout mice (Sgpl1 -/-).
Since then,several case studies have reported additional pathogenic variants and clinical manifestations in individuals with SPLIS.Close attention to the disease spectrum and the core manifestations of the disease (including PAI and SRNS),may lead to earlier identification of affected individuals and prevent irreversible outcomes,and/or allow participation in future clinical trials.Here we aim to review all patients reported to date including demographic,clinical,laboratory,and molecular aspects to allow a comprehensive overview of this rare disease.
Methods
Search strategies
We performed a systematic search of the PubMed,Web of Science,and Scopus Library databases for articles published in peer-reviewed journals up to July 2022.The keywords used in our search strategy were as follows:“sphingosine-1-phosphate lyase 1”,“SGPL1”,“SGPL1 deficiency”,“sphingosine-1-phosphate lyase insufficiency syndrome”,“SPL insufficiency syndrome”,“SPLIS”,“nephrotic syndrome type 14”,“NPHS14”.Manual screening for references from original articles and major reviews was performed to identify eligible studies.We adhered to the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) guideline,which is an evidence-based minimum set of items used for performing the systematic review [10] (Fig.1).
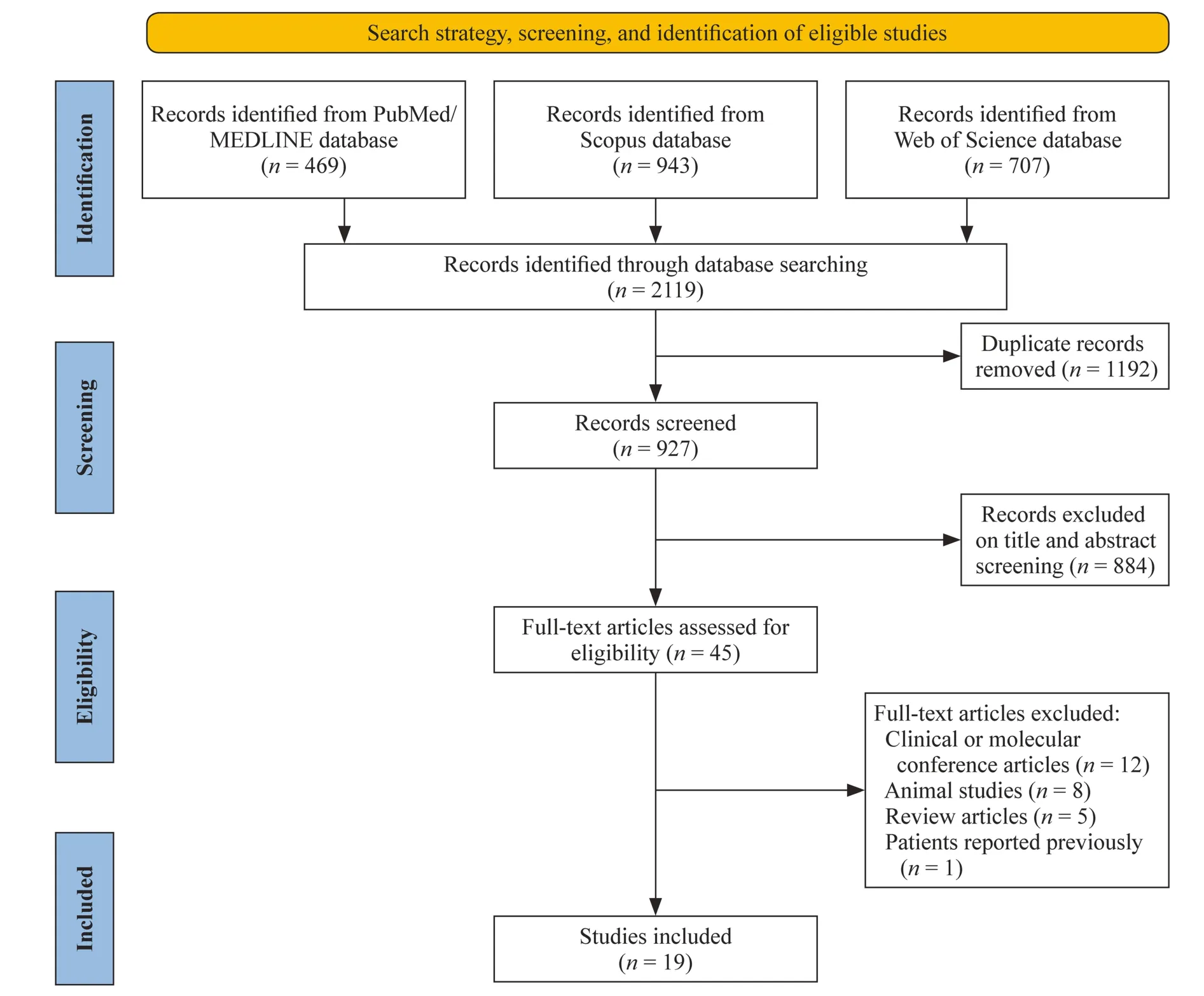
Fig.1 Preferred reporting items for systematic reviews and meta-analyses diagram illustrates study selection process
Study selection
The articles were first screened based on the title and abstract to exclude all irrelevant studies and were classified into three categories (include,exclude,or unclear);the full-text version of all unclear articles was checked and subsequently classified in one of the two categories (include or exclude).All full-text manuscripts were assessed for eligibility criteria:(1) written in English;(2) conducted on human subjects;and (3) report of at least one patient with SPLIS diagnosis.Studies using animal models,reviews,conference papers,and articles with previously reported patients were excluded.When necessary,the corresponding authors were contacted.
Data extraction
In the first step,two researchers extracted the data from all included studies based on the manuscript titles and abstracts.The following data were collected from all identified studies:(1) name of the first author;(2) publication year;(3) the number of participants;and (4) demographic,clinical,and paraclinical data.Two reviewers performed the selection process independently,while the third reviewer was consulted to resolve disagreements between two reviewers.
Statistical analysis
The statistical analyses were performed on data derived from related studies.Central and descriptive statistics were used for the interpretation of quantitative data.For variables with abnormal distribution,median and interquartile ranges (IQR) were calculated.All statistical tests were two-tailed,and aPvalue of less than 0.05 was considered statistically significant.The statistical analyses were performed using SPSS version 28.0 software (SPSS,Inc.,Chicago,IL,USA).
Results
We identified 55 patients from 19 previously published articles [7,9,11–27].The male/female ratio was 1.23 [28 male (54.9%) and 23 female (45.1%)] and for four patients the gender was unavailable.Most patients were of Turkish (13,23.6%),Arab (12,21.8%),Pakistani (7,12.7%),or North American (5,9%) origin.Most patients (39,70.9%) had consanguineous parents.A family history of fetal demise/early death,nephrotic syndrome,suspected immunodeficiency,or similar clinical phenotypes of the reported probands were reported in 29 (52.7%) patients.Most patients (54.9%,28 of 51) primarily manifested within the first year of life,while 27.5% (14 of 51) had prenatal evidence at antenatal paraclinical evaluation (including polyhydramnios,hydrops fetalis,hydrothorax and edema,borderline screening tests for trisomy 21,adrenal calcifications,and pulmonary hypoplasia),11.8% (6 of 51) presented between one and five years,and others (5.9%) in later ages (Supplementary Table 1).
There was a significant correlation between age at onset of symptoms and mortality rate (P< 0.002,Z=-3.1).All patients with a prenatal diagnosis of SPLIS died (P=0.003) at a median (IQR) age of 2 (1.4–5.3) months,while nearly half of patients (53.6%) with their first presentation in less than one year after birth had survived.The most common first clinical manifestations were kidney disorders (28,50.9%) [proteinuria (19,34.5%),edema (19,34.5%),hematuria (2,3.6%)] and neurological disorders (10,18.2%) [squint/ptosis (2,3.6%),hypotonia (2,3.6%),convulsion (2,3.6%),macrocephaly (1,1.8%),walking difficulty (2,3.6%)],followed by skin disorders (9,16.4%) [ichthyosis (4,7.3%),hyperpigmentation (7,12.7%)],hydrops (6,10.9%),endocrine-metabolic disorders (6,10.9%) [hypoglycemia (3,5.5%),adrenal crisis (2,3.6%),metabolic acidosis (2,3.6%)],urologic abnormalities (5,9.1%) [micropenis (5,9.1%),cryptorchidism (1,1.8%)],respiratory disorders (4,7.3%) [respiratory tract infection (2,3.6%),apnea (1,1.8%),pleural effusion (1,1.8%),hypoxia (1,1.8%)],failure to thrive (2,3.6%),and dysmorphia (2,3.6%).Hydrops and skin disorders as first presentation were associated with poor prognosis (P=0.01 andP=0.024,respectively).
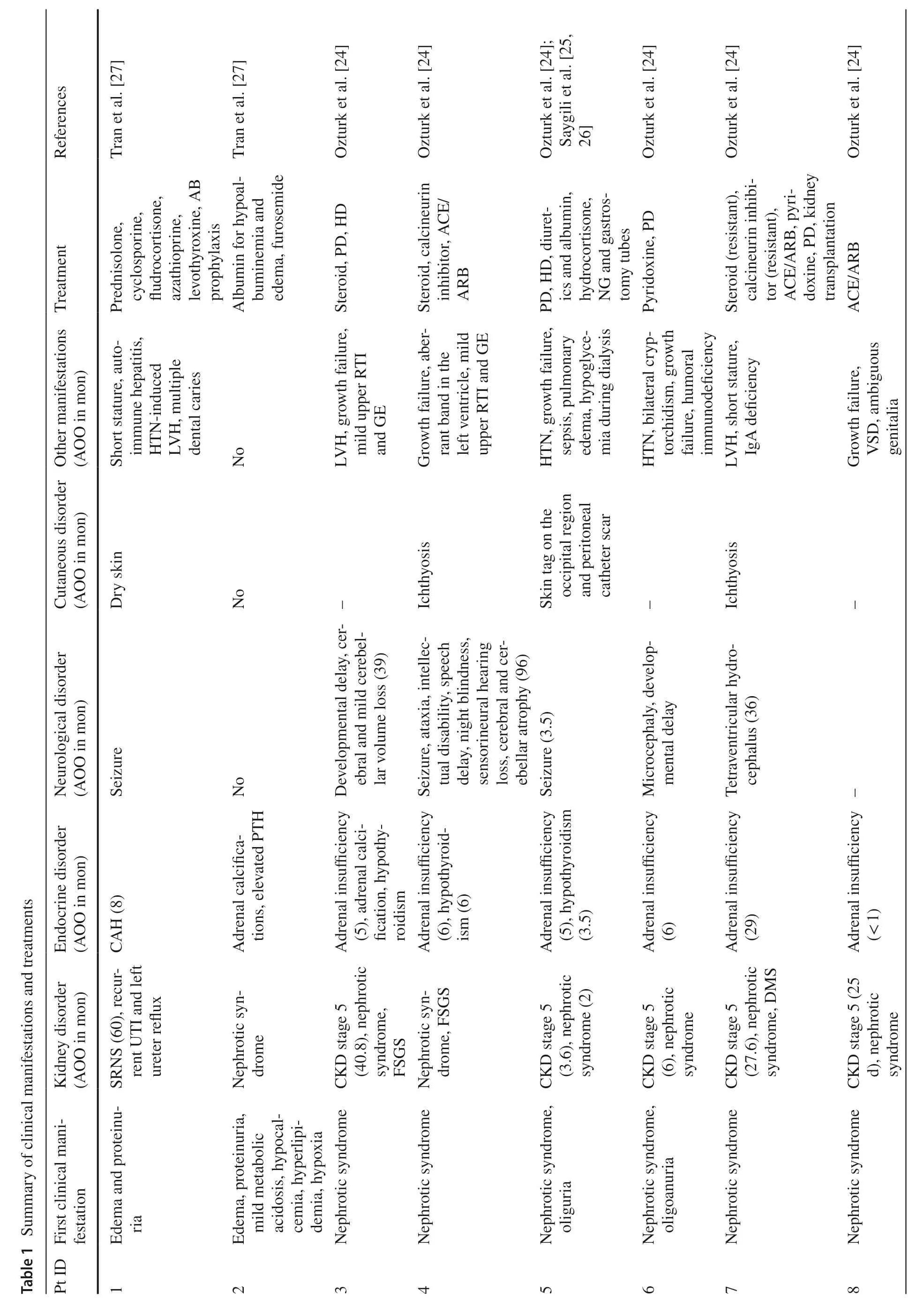
A summary of clinical manifestations reported in SPLIS patients is illustrated in Table 1.The most prevalent clinical feature was endocrine disorders (45,86.5%).PAI as the predominant endocrinopathy in 37 (71.2%) patients,was diagnosed at a median (IQR) age of 21.5 (5.8–30.8) months with the oldest presenting at 11 years of age.Four additional patients had adrenal calcifications but no reported PAI.The vast majority of those with PAI also had nephrotic syndrome (30 of 37,81.1%).The second most frequent endocrinopathy was hypothyroidism,either primary (17,30.9%) or due to suppression of the thyroid gland by biotin (1,1.8%).Secondary or tertiary hyperparathyroidism was reported in 4 (7.7%) patients.Primary gonadal failure was reported in almost one-third of male patients (9,32.1%),presenting features included micropenis and cryptorchidism.Kidney disorders (42,80.8%),mainly in the form of SRNS,were the second most common clinical characteristic,presenting at a median (IQR) age of 4 (1–33) months.Nineteen (36.5%) patients with SRNS had developed end-stage kidney disease (ESKD) at a median (IQR) age of 6 (1.4–42.6) months.Neurological disorders (30,57.7%) mainly included developmental delay/regression (12,23.1%),sensorineural hearing loss (10,19.2%),ophthalmopathies [strabismus (6,10.9%),ptosis (3,5.5%),retinopathy (2,3.6%),oculomotor palsy (1,1.8%) and cortical visual impairment (1,1.8%)],convulsion (9,17.3%),peripheral neuropathy (8,15.4%),hypotonia (6,11.5%),and microcephaly (6,11.5%).Skin disorders including ichthyosis/hyperkeratosis (20,38.5%) and hyperpigmentation secondary to adrenal insufficiency (13,25%) were observed in half of the patients (27,51.9%).
Immunologic defects were reported in 14 SPLIS patients (Supplementary Table 2).Lymphopenia was reported in 24 out of 35 (68.6%) patients with available cell blood count.In twelve patients flow cytometric alterations including low CD3+T cells (9 of 11,81.8%),low CD4+T cells (8 of 12,66.7%),low CD8+T cells (9 of 12,75.0%),low CD19+B cells (7 of 11,63.6%),and low CD16+56+natural killer cells (3 of 12,25.0%) were observed.Low serum immunoglobulins including low immunoglobulin (Ig) G (5 of 8,62.5%),IgA (2 of 6,33.3%),and IgM (1 of 5,20%).Absent or low T-cell receptor excision circles indicative of recent thymic emigrants (a recommended uniform screening panel test for severe combined immunodeficiency) were reported in three patients.
Overall,there were 30 differentSGPL1mutations identified in the 55 patients,including 21 missense,seven frameshift,two splice-site,two in Del,and one nonsense variant in homozygous or compound heterozygosity (Supplementary Fig.1,Supplementary Table 3).The most common genetic variant in 11 (20%) patients was c.665G > A (p.Arg222Gln).Mutations were located throughoutSGPL1,and there did not appear to be any mutational hot spots.However,the pyridoxal-dependent decarboxylase conserved domain had a large proportion of the mutations.
Twenty-six (49.1%) patients with available outcomes were deceased at a median (IQR) age of 5 (1.5–30.5) months.Four (15.4%) patients were lost due to fetal demise,while other born patients died following ESKD (6,23%),sepsis/septic shock (6,23%),pulmonary edema/respiratory failure (3,11.5%),hypovolemic shock (1,3.8%),congestive heart failure and bradyarrhythmia (1,3.8%),and cerebral edema (1,3.8%),and sudden death with unknown cause (2,7.7%).Among all clinical manifestations,a significant correlation was identified between mortality risk and the development of nephrotic syndrome (P=0.014) and adrenal calcification (P=0.007).In addition,lower age at onset of nephrotic syndrome and endocrinopathies were associated with a higher mortality rate (P< 0.001 andP=0.008,respectively).There was no significant correlation between mortality rate and lymphopenia (P=0.088),neurologic manifestations [including hypotonia (P=0.395),developmental delay/regression (P=0.750),convulsion (P=0.715),sensorineural hearing loss (P=1.0),chorea (P=0.460),ataxia (P=0.614),peripheral neuropathy (P=0.674),encephalitis (P=1.0),microcephaly (P=0.082),macrocephaly (P=1.0)],endocrinopathies [adrenal insuffi-ciency (P=0.496),hypothyroidism (P=0.108),hyperparathyroidism (P=0.322),hypogonadism (P=0.062)],ichthyosis (P=0.064),and the type of presenting symptom (other than hydrops and skin disorders).
Discussion
In a systematic search of the literature for patients with SPLIS,we found 55 patients and reviewed epidemiologic features and clinical,molecular,and immunological characteristics.A high rate of positive family history follows the autosomal recessive pattern of SPLIS,further confirmed by a
higher prevalence of the disorder in countries with high rates of consanguineous marriages.This emphasizes the importance of documenting a precise family history,which can provide clues for suspecting autosomal recessive diseases such as SPLIS.A negative family history,however,does not preclude the diagnosis.Detection of prenatal evidence on fetal paraclinical screening and younger age at the first presentation of SPLIS are associated with unfavorable prognosis.A previous study has shown that lower age at diagnosis of adrenal insufficiency and kidney dysfunction is associated with lower survival time [3].This correlation may be explained by the higher severity of disorder among patients initially manifesting in the prenatal period or perinatally,emphasizing the necessity for timely molecular and biochemical screening ofSGPL1defects in suspected patients.
Variable combinations of organ involvement make the diagnosis of SPLIS challenging.Although the overall most common components of SPLIS are related to endocrinopathies,kidney disorders mainly in the form of nephrotic syndrome,are the first manifestations to appear in a median (IQR) age of 5 (1–33) months.Renal tubular dilatation,effacement of podocytes,expansion of mesangial matrix,and glomerulosclerosis are the most common histological findings following a kidney biopsy.Although most patients are given the diagnosis of focal segmental glomerulosclerosis,histopathological studies may not be straightforward in some patients.The development of nephrotic syndrome has a negative impact on the mortality rate.Unfortunately,the rapid progression to ESKD occurs in more than onethird of patients with nephrotic syndrome and was the main reason for death in 23% of all patients [18].In other patients,nephropathy and ESRD may develop many years after the initial presenting feature.
Endocrinopathies reported in SPLIS have a wide spectrum which is unique to SPLIS in comparison to other sphingolipidoses.Adrenal insufficiency and/or adrenal calcification were the predominant endocrine disorder presenting in 74.5% of patients with nephrotic syndrome.However,this may be an underestimation,particularly in patients diagnosed with isolated nephrotic syndrome,due to invariable presentations of adrenal insufficiency which may be masked by early initiation of steroid therapy for the nephrotic syndrome [28].In male patients,physical examination to identify micropenis or cryptorchidism or serum evaluation of sexual hormones should be performed as primary gonadal failure was reported in almost one-third of male patients and was significantly correlated with mortality rate.
Neurological disorders,as the third most common characteristic of SPLIS,mainly included developmental delay/regression,sensorineural hearing loss,convulsion,peripheral neuropathy,hypotonia,cranial nerve palsies,and microcephaly,the latter of which was correlated with mortality rate.S1P metabolism plays an important role in neurological pathways,including presynaptic architecture and neuronal autophagy [29,30].In addition,hyperphosphorylated tau protein and increased histone acetylation have been found in primary cultured neurons and astrocytes,respectively [31],which can be proposed as the underlying neuropathology mechanism.
In laboratory evaluation,in addition to the expected increase in S1P,serum and liver sphingolipids (including sphingosine,ceramide,and sphingomyelin) have been shown to be increased inSgpl1 -/-mice which can contribute to disturbances in lipid homeostasis and can explain some of the metabolic abnormalities [32].Interestingly,among metabolic parameters,the plasma S1P/dihydroS1P ratio can be utilized to help confirmation of clinically diagnosed patients [19].Another laboratory clue for SPLIS diagnosis would be lymphopenia reported in almost 68.6% of patients,along with disturbed percentages of lymphocyte subsets and immunoglobulins compared to the normal range for age.Although low immunologic parameters may be attributed to immunosuppressive agents by physicians,studies onS1pl -/-mice have demonstrated lymphopenia,with sequestration of mature T cells in the thymus and peripheral lymphoid organs,and lethal non-lymphoid lesions [33].Weber et al.argued that accumulation of ceramides increases thymocyte apoptosis and depletion of thymic early T cell progenitors,abrogating postnatal thymocyte development [34].However,the main mechanism for lymphopenia is a block in lymphocyte egress due to the failure of S1P lyase to generate the S1P chemotactic gradient [35,36].This is consistent with the finding that most SPLIS patients retain some degree of T cell function.While low immunoglobulin levels may be attributed to the presence of SRNS,it remains to be determined whether B lymphocyte dysfunction is a component of the immune features of SPLIS.
Treatment modalities for SPLIS were previously limited to supportive care,however,since the diagnosis of the first patient in 2017,major efforts have been made to provide a curative option for affected individuals including mutantSGPL1repair,enzymatic replenishment,augmentation of patients’ enzyme activity,or bone marrow transplantation [37].Recently,Zhao et al.have introduced vitamin B6 cofactor supplementation as a potential targeted therapy for SPLIS.They investigated lymphocyte counts and plasma S1P levels before and after administration of vitamin B6 in four SPLIS patients and in two of them found resolution of lymphopenia and also augmentation of S1P lyase protein and activity and reduced sphingolipids in patients’ fibroblasts.However,they demonstrated vitamin B6 supplementation may only benefit patients with susceptibleSGPL1missense mutations (such as patients homozygous for p.Arg222Gln or p.Gly360Val or compound heterozygous for p.Phe290Leu and p.Tyr331*) and declined functional activity [3].In addition,there is no clear evidence on the benefit of vitamin B6 in the prevention of end-organ damage,although murine studies have shown that even small amounts of S1P lyase activity can remarkably prevent or reduce kidney,endocrine,and neurological complications [33].Enzyme therapy might also be challenging because S1P lyase is attached to the outer membrane of the endoplasmic retinaculum and not in the lysosome [3].As a promising future treatment,gene therapy using the AAV9 vector to transfer normalSGPL1intoS1pl -/-mice has been associated with improved survival [38].
Taken together,SPLIS is a novel sphingolipidosis with heterogenous clinical features.Patients may be diagnosed as having an isolated nephrotic syndrome or endocrine disorder for years before a molecular genetic diagnosis ofSGPL1mutations is made.In view of the reports of isolated adrenal or proteinuric kidney disease at the presentation it would be pertinent forSGPL1to be added to diagnostic next-generation sequencing genetic panels and virtual gene panels following whole exome and whole genome sequencing for patients with PAI or SRNS,or a combination of these phenotypes.Patients with milder forms of SPLIS may easily go undiagnosed or misdiagnosed,so identification of a family history of SRNS or PAI in an individual with any of the main features of SPLIS should raise suspicion and prompt genetic diagnostic testing.
Supplementary InformationThe online version contains supplementary material available at https:// doi.org/ 10.1007/ s12519-022-00615-4.
Author contributionsPZ contributed to conceptualization,methodology and data curation.MA,CZ,and TP contributed to reviewing and editing.SJA and SJD contributed to investigation,reviewing and editing.NF contributed to investigation.JM contributed to conceptualization,methodology,data curation,investigation,formal analysis,supervision,visualization,and original draft preparation.All the authors approved the final version of the manuscript.
FundingNone.
Data availabilityAll data generated or analyzed during this study are included in this published article and its supplementary information files.
Declarations
Ethical approval Ethical approval was not necessary for the performance of this review,as it involved secondary review of existing literature.
Conflict of interestNo financial or non-financial benefits have been received or will be received from any party related directly or indirectly to the subject of this article.The authors have no conflict of interest to declare.
 World Journal of Pediatrics2023年5期
World Journal of Pediatrics2023年5期
- World Journal of Pediatrics的其它文章
- Multigenerational birth cohort study in China:importance,necessity and beyond
- Biliatresone:progress in biliary atresia study
- Early recombinant human growth hormone treatment improves mental development and alleviates deterioration of motor function in infants and young children with Prader–Willi syndrome
- Nusinersen for spinal muscular atrophy types II and III:a retrospective single-center study in South Korea
- Relationship between early nutrition and deep gray matter and lateral ventricular volumes of preterm infants at term-equivalent age
- Impact of the COVID-19 kindergarten closure on overweight and obesity among 3-to 7-year-old children